


2020

1st October 2023, Caesium-137
On April 26, 1986, reactor 4 at the Chernobyl nuclear power plant in Pripyat, Ukraine suffered catastrophic meltdown and exploded, killing 2 workers and unleashing large amount of radiation across the European continent. In the 3 months that followed, 28 plant workers and first responders died from acute radiation sickness. Wildlife was also affected, and restrictions were put in place to prevent people from eating radioactively contaminated meat [1].Since then, the contamination of deer and roe deer has decreased over time. However, the radioactivity seen in wild boars stayed surprisingly high, well above the regulatory food limit. For many years scientists assumed that the high radioactivity seen in wild boars was the result of the Chernobyl disaster. However, a team of scientists have now determined the exact cause of the radiation. Their study published in the journal Environmental Science & Technology, looked at the levels of caesium-137 (CHEBI:196959), a key radioactive isotope in wild boar samples. Caesium-137 has a half-life of around 30 years, hence after 30 years, half of the material should have decayed by itself. However, in Bavarian boar flesh, radiation levels have remained almost constant after nearly 37 years since the Chernobyl disaster [2,3].
A team led by Professor Georg Steinhauser at TU Wien decided to investigate the origin as well as the amount of radioactivity in boars. They worked with hunters who collected wild boar meat across 11 Bavarian districts across southern Germany. The team then measured caesium levels using γ-ray spectroscopy. It is well-known that radioactive caesium can result from both nuclear weapons explosion and nuclear energy production. The element comes in different isotopic composition, caesium-135 and caesium-137, depending on the source. From previous studies, it was known that a higher ratio of caesium-137 to caesium-135 indicated a nuclear weapons explosion and a lower ratio is linked with nuclear reactors. By analysing the ratio of the isotopes of caesium, the team were able to narrow down the source of the radiation and found that the boars bore the marks of a different period — nuclear weapons testing of the 1960s [4].
The study showed that caesium-137 from the Chernobyl disaster remained the most significant contributor to wild boar contamination. However, about 25% of wild boar samples exhibited significant contribution from caesium-137 produced as a result of atmospheric weapons fallout. It is suggested that wild boars probably ingested the caesium from contaminated truffle mushrooms. The study illustrates that nuclear weapon tests carried out 60 years ago, still impact remote natural environments, wildlife, and food security today [4].
The background image is a detail from a Creative Commons licensed photograph of a wild boar in the Bavarian forest national park.
Reference(s)
- Scientists finally know why Germany's wild boar are surprisingly radioactive (The Washington Post press release, 3 September 2023).
- Germany's radioactive boars are a bristly reminder of nuclear fallout (Science press release, 30 August 2023).
- Chernobyl: Scientists solve mystery of why wild boars are more radioactive than other animals (euronews press release, 2 September 2023).
- Stäger, F., Zok, D., Schiller, A.K., Feng, B. and Steinhauser, G. (2023) Disproportionately high contributions of 60 year old weapons-137Cs explain the persistence of radioactive contamination in Bavarian wild boars. Environ Sci Technol., Epub ahead of print.
1st September 2023, Caesium-137
On April 26, 1986, reactor 4 at the Chernobyl nuclear power plant in Pripyat, Ukraine suffered catastrophic meltdown and exploded, killing 2 workers and unleashing large amount of radiation across the European continent. In the 3 months that followed, 28 plant workers and first responders died from acute radiation sickness. Wildlife was also affected, and restrictions were put in place to prevent people from eating radioactively contaminated meat [1].Since then, the contamination of deer and roe deer has decreased over time. However, the radioactivity seen in wild boars stayed surprisingly high, well above the regulatory food limit. For many years scientists assumed that the high radioactivity seen in wild boars was the result of the Chernobyl disaster. However, a team of scientists have now determined the exact cause of the radiation. Their study published in the journal Environmental Science & Technology, looked at the levels of caesium-137 (CHEBI:196959), a key radioactive isotope in wild boar samples. Caesium-137 has a half-life of around 30 years, hence after 30 years, half of the material should have decayed by itself. However, in Bavarian boar flesh, radiation levels have remained almost constant after nearly 37 years since the Chernobyl disaster [2,3].
A team led by Professor Georg Steinhauser at TU Wien decided to investigate the origin as well as the amount of radioactivity in boars. They worked with hunters who collected wild boar meat across 11 Bavarian districts across southern Germany. The team then measured caesium levels using γ-ray spectroscopy. It is well-known that radioactive caesium can result from both nuclear weapons explosion and nuclear energy production. The element comes in different isotopic composition, caesium-135 and caesium-137, depending on the source. From previous studies, it was known that a higher ratio of caesium-137 to caesium-135 indicated a nuclear weapons explosion and a lower ratio is linked with nuclear reactors. By analysing the ratio of the isotopes of caesium, the team were able to narrow down the source of the radiation and found that the boars bore the marks of a different period — nuclear weapons testing of the 1960s [4].
The study showed that caesium-137 from the Chernobyl disaster remained the most significant contributor to wild boar contamination. However, about 25% of wild boar samples exhibited significant contribution from caesium-137 produced as a result of atmospheric weapons fallout. It is suggested that wild boars probably ingested the caesium from contaminated truffle mushrooms. The study illustrates that nuclear weapon tests carried out 60 years ago, still impact remote natural environments, wildlife, and food security today [4].
The background image is a detail from a Creative Commons licensed photograph of a wild boar in the Bavarian forest national park.
Reference(s)
- Scientists finally know why Germany's wild boar are surprisingly radioactive (The Washington Post press release, 3 September 2023).
- Germany's radioactive boars are a bristly reminder of nuclear fallout (Science press release, 30 August 2023).
- Chernobyl: Scientists solve mystery of why wild boars are more radioactive than other animals (euronews press release, 2 September 2023).
- Stäger, F., Zok, D., Schiller, A.K., Feng, B. and Steinhauser, G. (2023) Disproportionately high contributions of 60 year old weapons-137Cs explain the persistence of radioactive contamination in Bavarian wild boars. Environ Sci Technol., Epub ahead of print.

???en_GB.entity.month.date.224???, ???en_GB.entity.month.title.224???
???en_GB.entity.month.summary.224??? ???en_GB.entity.month.content.224???Reference(s)
-
???en_GB.entity.month.references.224???

6th July 2023, Cannabidiol
It is pretty disappointing when you get home and find that the box of freshly, sweet strawberries that you recently picked from the supermarket have started to rot. Recently, a team of researchers at Thammasat University in Thailand have incorporated cannabidiol (also known as CBD, CHEBI:69478) into an edible antimicrobial coating to delay the rotting process and increase the strawberries' shelf life [1]. CBD, a non-psychoactive cannabinoid found in the cannabis plant has many potential therapeutic properties including antioxidant and antimicrobial properties. In previous studies, it has been shown to inhibit the growth of certain bacteria and pathogenic fungi such as the ones that cause fresh fruits and vegetables to rot [2].Pongpat and colleagues wanted to check whether a food coating made using CBD-filled nanoparticles could promote antimicrobial activity and extend the freshness of strawberries. Their work published in the journal ACS Applied Materials & Interfaces encapsulated CBD in a widely used biodegradable polymer in drug delivery known as poly(ᴅ,ʟ-lactide-co-glycolide). The team mixed the most stable nanoparticles containing 20% by weight CBD with sodium alginate in water. Strawberries were subsequently submerged in a solution containing different amounts of nanoparticles and then dipped into a mixture of ascorbic acid and calcium chloride which turned the colourless coating into a gel [3].
Untreated and treated strawberries were placed in open plastic containers at refrigerator temperatures for 15 days and monitored. It was found that strawberries treated with CBD ripened and decayed much more slowly compared to untreated samples, most likely due to reduced microbial growth. Furthermore, coatings with the most CBD-loaded nanoparticles had a greater effect in enhancing the strawberries' antioxidant activity and prolonged quality by preserving their dark red appearance. They also exhibited a high antimicrobial protection over the storage period and increased the shelf life of strawberries. The results of this study demonstrate how encapsulated CBD can potentially be used as an efficient active food coating agent [3].
The background image is a detail from a Creative Commons licensed photograph of fresh strawberries being sold in plastic containers.
Reference(s)
- An edible CBD coating could extend the shelf life of strawberries (Phys.Org press release, 17th May 2023).
- Extending the shelf-life of berries with CPD (Food manufacture press release, 19th May 2023).
- Sukhavattanakul, P., Thanyacharoen, T., Chuysinuan, P., Techasakul, S. and Ummartyotin, S. (2023) Influence of a transparent and edible coating of encapsulated cannabidiol nanoparticles on the quality and shelf life of strawberries. ACS Appl Mater Interfaces, 15(19), 23834–23843.
7th June 2023, Cannabidiol
It is pretty disappointing when you get home and find that the box of freshly, sweet strawberries that you recently picked from the supermarket have started to rot. Recently, a team of researchers at Thammasat University in Thailand have incorporated cannabidiol (also known as CBD, CHEBI:69478) into an edible antimicrobial coating to delay the rotting process and increase the strawberries' shelf life [1]. CBD, a non-psychoactive cannabinoid found in the cannabis plant has many potential therapeutic properties including antioxidant and antimicrobial properties. In previous studies, it has been shown to inhibit the growth of certain bacteria and pathogenic fungi such as the ones that cause fresh fruits and vegetables to rot [2].Pongpat and colleagues wanted to check whether a food coating made using CBD-filled nanoparticles could promote antimicrobial activity and extend the freshness of strawberries. Their work published in the journal ACS Applied Materials & Interfaces encapsulated CBD in a widely used biodegradable polymer in drug delivery known as poly(ᴅ,ʟ-lactide-co-glycolide). The team mixed the most stable nanoparticles containing 20% by weight CBD with sodium alginate in water. Strawberries were subsequently submerged in a solution containing different amounts of nanoparticles and then dipped into a mixture of ascorbic acid and calcium chloride which turned the colourless coating into a gel [3].
Untreated and treated strawberries were placed in open plastic containers at refrigerator temperatures for 15 days and monitored. It was found that strawberries treated with CBD ripened and decayed much more slowly compared to untreated samples, most likely due to reduced microbial growth. Furthermore, coatings with the most CBD-loaded nanoparticles had a greater effect in enhancing the strawberries' antioxidant activity and prolonged quality by preserving their dark red appearance. They also exhibited a high antimicrobial protection over the storage period and increased the shelf life of strawberries. The results of this study demonstrate how encapsulated CBD can potentially be used as an efficient active food coating agent [3].
The background image is a detail from a Creative Commons licensed photograph of fresh strawberries being sold in plastic containers.
Reference(s)
- An edible CBD coating could extend the shelf life of strawberries (Phys.Org press release, 17th May 2023).
- Extending the shelf-life of berries with CPD (Food manufacture press release, 19th May 2023).
- Sukhavattanakul, P., Thanyacharoen, T., Chuysinuan, P., Techasakul, S. and Ummartyotin, S. (2023) Influence of a transparent and edible coating of encapsulated cannabidiol nanoparticles on the quality and shelf life of strawberries. ACS Appl Mater Interfaces, 15(19), 23834–23843.

8th May 2023, 1,2,3-cyclohexatriene
There has been a long interest in compounds with the molecular formula C6H6 especially benzene which has been subjected to rigorous scientific investigation. Of these, the high energy compound, 1,2,3-cyclohexatriene (CHEBI:195206) has been relatively ignored. Compared to the alternating conjugated double bonds of benzene, the three contiguous double bonds in 1,2,3-cyclohexatriene lack any stabilising aromaticity and its strained ring structure, led a few to believe this benzene isomer was simply too unstable to be synthetically useful [1,2].However, a team of chemists at the University of California, Los Angeles in the US, recently identified the compound to be a versatile reagent in organic synthesis. Their study published in the journal Nature used a method developed in the 1990s to generate reactive 1,2,3-cyclohexatriene intermediates from a precursor, probing the reaction scope by introducing a variety of trapping agents which were subsequently used to produce a diverse array of fused-ring adducts [3].
The team demonstrated that 1,2,3-cyclohexatriene and its derivatives participate in a diverse range of cycloaddition, nucleophilic addition and σ-bond insertion reactions, enabling them to synthesise stereochemically and topologically complex molecules in just a few steps. DFT studies in conjunction with experimental studies showed that 1,2,3-cyclohexatriene derivatives could be used in certain reactions regardless of their high degree of reactivity and short lifetime. To date, derivatives of 1,2,3-cylohexatriene are largely unexplored and the team hopes that their study will inspire other researchers to explore their synthetic potential in the synthesis of important compounds [3].
The background image is a detail from a Creative Commons licensed photograph of Royce Hall at UCLA.
Reference(s)
- Benzene's forgotten isomer takes centre stage in organic synthesis (Chemistry World press release, 2 May 2023).
- A fresh look at 1,2,3-cyclohexatriene shows it could be used as a versatile reagent in organic synthesis (Phys.Org press release, 3 May 2023).
- Kelleghan, A.V., Bulger, A.S., Witkowski, D.C. and Garg, N.K. (2023) Strain-promoted reactions of 1,2,3-cyclohexatriene and its derivatives. Nature, Epub ahead of print.
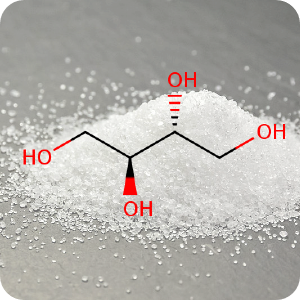
11th April 2023, Erythritol
In the past few decades artificial sweeteners have been widely introduced into the food chain to help reduce insulin resistance and obesity. They are common ingredients in processed foods, soft drinks and personal care products and generally considered safe by regulatory agencies such as the FDA [1]. Patients with type 2 diabetes and obesity are frequently advised that the use of artificial sweeteners as a sugar substitute can improve glycaemic control and help achieve weight loss. However, little is known about their long-term health effects in humans [2].Erythritol (CHEBI:17113) is a four-carbon polyol commonly used as a sugar substitute. It has a moderate sweetening intensity and lower calorie content than sucrose (table sugar), and its market share is predicted to double within the sweetener sector in the next 5 years due to its popularity. In nature, it is present in low amounts in fruits and vegetables and when ingested, it is poorly metabolised by the human body and most of it is excreted unmodified via the urine without changing blood glucose and insulin levels. For these reasons, it is consequently characterised as both a 'zero-calorie' and 'natural' sweetener. However, when erythritol is incorporated into processed foods, it is added at levels 1000-fold higher than endogenous levels [3].
A recent study published in the journal Nature Medicine highlighted the link between erythritol and increased risk of heart attack, stroke and death. The study conducted by researchers at the Cleveland Clinic looked at a 4,000-patient cohort in the United States and Europe who were undergoing elective cardiac evaluation. They found that patients who had greater levels of erythritol in their blood had a higher chance of experiencing adverse cardiac events. In preclinical studies, they found that ingestion of erythritol increased blood clot formation [3]. Researchers caution that further studies are needed into the health effects of erythritol and other sweeteners which they did not study, and that participants involved in the study generally had a high prevalence of cardiovascular disease, hence the 'translatability' of these findings to the general population needs to be determined. However, the results still pose a significant challenge to 'misleading marketing' that pitches erythritol as a healthy, natural sugar alternative [1].
The background image is a detail from a Creative Commons licensed photograph of erythritol crystals.
Reference(s)
- Artificial sweetener linked to higher heart attack risk, study says (The Washington Post press release, 28 February 2023).
- Pang, M.D., Goossens, G.H. and Blaak, E.E. (2021) The impact of artificial sweeteners on body weight control and glucose homeostasis. Front Nutr., 7, 598340.
- Witkowski, M., Nemet, I., Alamri, H., Wilcox, J., Gupta, N., Nimer, N., Haghikia, A., Li, X.S., Wu, Y., Saha, P.P., Demuth, I., König, M., Steinhagen-Thiessen, E., Cajka, T., Fiehn, O., Landmesser, U., Tang, W.H.W. and Hazen, S.L. (2023) The artificial sweetener erythritol and cardiovascular event risk. Nat Med., Epub ahead of print.
7th March 2023, Erythritol
In the past few decades artificial sweeteners have been widely introduced into the food chain to help reduce insulin resistance and obesity. They are common ingredients in processed foods, soft drinks and personal care products and generally considered safe by regulatory agencies such as the FDA [1]. Patients with type 2 diabetes and obesity are frequently advised that the use of artificial sweeteners as a sugar substitute can improve glycaemic control and help achieve weight loss. However, little is known about their long-term health effects in humans [2].Erythritol (CHEBI:17113) is a four-carbon polyol commonly used as a sugar substitute. It has a moderate sweetening intensity and lower calorie content than sucrose (table sugar), and its market share is predicted to double within the sweetener sector in the next 5 years due to its popularity. In nature, it is present in low amounts in fruits and vegetables and when ingested, it is poorly metabolised by the human body and most of it is excreted unmodified via the urine without changing blood glucose and insulin levels. For these reasons, it is consequently characterised as both a 'zero-calorie' and 'natural' sweetener. However, when erythritol is incorporated into processed foods, it is added at levels 1000-fold higher than endogenous levels [3].
A recent study published in the journal Nature Medicine highlighted the link between erythritol and increased risk of heart attack, stroke and death. The study conducted by researchers at the Cleveland Clinic looked at a 4,000-patient cohort in the United States and Europe who were undergoing elective cardiac evaluation. They found that patients who had greater levels of erythritol in their blood had a higher chance of experiencing adverse cardiac events. In preclinical studies, they found that ingestion of erythritol increased blood clot formation [3]. Researchers caution that further studies are needed into the health effects of erythritol and other sweeteners which they did not study, and that participants involved in the study generally had a high prevalence of cardiovascular disease, hence the 'translatability' of these findings to the general population needs to be determined. However, the results still pose a significant challenge to 'misleading marketing' that pitches erythritol as a healthy, natural sugar alternative [1].
The background image is a detail from a Creative Commons licensed photograph of erythritol crystals.
Reference(s)
- Artificial sweetener linked to higher heart attack risk, study says (The Washington Post press release, 28 February 2023).
- Pang, M.D., Goossens, G.H. and Blaak, E.E. (2021) The impact of artificial sweeteners on body weight control and glucose homeostasis. Front Nutr., 7, 598340.
- Witkowski, M., Nemet, I., Alamri, H., Wilcox, J., Gupta, N., Nimer, N., Haghikia, A., Li, X.S., Wu, Y., Saha, P.P., Demuth, I., König, M., Steinhagen-Thiessen, E., Cajka, T., Fiehn, O., Landmesser, U., Tang, W.H.W. and Hazen, S.L. (2023) The artificial sweetener erythritol and cardiovascular event risk. Nat Med., Epub ahead of print.
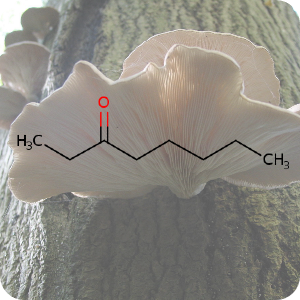
1st February 2023, 3-octanone
Oyster mushrooms (Pleurotus ostreatus) are creamy grey edible mushrooms that have a taste reminiscent of anise, and often served at high-end restaurants. In their natural environment, oyster mushrooms are carnivorous — they kill and consume nematodes. Prior research has shown that oyster mushrooms are able to kill their prey by producing potent toxins that paralyze nematodes within a few minutes of contact. To date, the nematicidal compound produced by the oyster mushrooms remained inconclusive [1,2].A team of researchers from Taiwan have recently used gas chromatography-mass spectrometry to identify the toxic compound emitted by oyster mushrooms for killing prey. Their paper published in journal Science Advances, identified 3-octanone (CHEBI:80946), a volatile ketone in the mushroom's toxocysts [3]. The chemical is widely present in the volatiles of many plants indicating that this natural ketone might protect plants from herbivorous insects [4].
The toxin triggers paralysis and elicits massive calcium influx and cell necrosis throughout the nematode neuromuscular system. The mitochondria are punctured and fragmented. The toxin also disrupts the integrity of cell membranes leading to widespread cell death. The team observed that a high concentration (>50%) of 3-octanone was required to paralyze and kill the nematodes. Structurally related compounds were also tested on nematodes and shown to be biotoxic, with the length of the ketone carbon chain being critical for nematocidal activity [3].
The background image is a detail from a Creative Commons licensed photograph of the fungus, Pleurotus ostreatus.
Reference(s)
- 3-octanone identified as the toxic agent used by oyster mushrooms to kill prey (Phys.org press release, 19 January 2023).
- Barron, G.L. and Thorn, R.G. (1987) Destruction of nematodes by species of Pleurotus. Can. J. Bot., 65, 774–778.
- Lee, C.H., Lee, Y.Y., Chang, Y.C., Pon, W.L., Lee, S.P., Wali, N., Nakazawa, T., Honda, Y., Shie, J.J. and Hsueh, Y.P. (2023) A carnivorous mushroom paralyzes and kills nematodes via a volatile ketone. Sci Adv., 9(3), eade4809.
- Koedam, A. and Gijbels, M.J.M. (1978) Isolation and identification of 3-octanone in the essential oil of Rosmarinus officinalis L. Z. Naturforsch., 33c, 144–145.
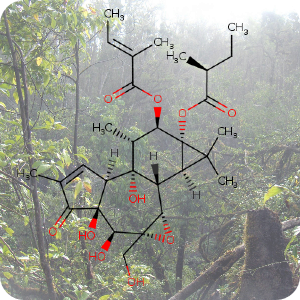
1st January 2023, Tigilanol tiglate
Researchers at Stanford university have figured out a sustainable and reliable way to synthesise a promising plant-derived anticancer compound. To date, the compound appeared synthetically innacessible given its complex structure and no plausible path seemed to exist for synthesising it practically in a laboratory. The compound known as tigilanol tiglate (designated EBC-46, CHEBI:194146) was initially identified by an automated drug candidate screening process by the Australian company QBiotics [1].In nature, tigilanol tiglate is found in the seeds of the pink fruit of the blushwood tree, Fontainea picrosperma. It is proposed that tigilanol tiglate induces rapid tumour ablation, in part by an isoform-selective modulation of protein kinase C (PKC) [2]. After intratumoural administration, the compound induces a localized immune response and rupture of tumour vasculature, leading to haemorrhagic necrosis, rapid tumour cell death and subsequent clearance of the solid tumour [3]. In 2020, the European Medicines Agency and the Food and Drug Administration approved a tigilanol tiglate-based medication, sold under the brand name Stelfonta for the treatment of non-metastatic mast cell tumours in canines [4,5]. A recent clinical study showed a 75% cure rate in canines after a single intratumoural injection and an 88% remission after a second dose. Clinical trials have since commenced in human patients for the treatment of a broad range of cancers including head and neck, melanoma, and soft tissue cancers [6].
The only source of tigilanol tiglate is a tree found in a small rainforest region of Northeastern Australia. The tree's limited numbers and restricted distribution prompted consideration of designed tree plantations to address supply needs. However, this presents further challenges, for example the trees require pollination, meaning the right sort of pollinating animals must be present, and the trees are affected by seasonal and climate variations [7]. Given the immediate clinical and research value of tigilanol tiglate, Wender and colleagues at Stanford university proposed a semisynthetic route from a more readily available precursor [8].
Their research work published in the journal Nature Chemistry proposed phorbol, a plant derived compound as starting point for synthesising tigilanol tiglate [8]. Globally, around 7,000 plant species produce phorbol derivatives and phorbol-rich seeds are commercially inexpensive. The team extracted phorbol-rich oil from the seeds of Croton tiglium, a herb used in traditional Chinese medicine. After processing the oil to yield phorbol, they then had to overcome the previously insurmountable challenge of bedecking a part of the molecule, called the B ring, with carefully placed oxygen atoms that were critical for PKC activity. The team succeeded in adding extra oxygen atoms to phorbol's B ring via an ene reaction conducted under flow conditions. The team then introduced other functional groups in a stepwise, controlled manner to obtain the required spatial arrangements of the atoms. In total, 12 steps were required to produce tigilanol tiglate itself and 4-6 steps to obtain derivatives of tigilanol tiglate. It is hoped that the broader availability of tigilanol tiglate and its derivatives afforded by this approach will accelerate research into potentially new treatments [8].
The background image is a detail from a Creative Commons licensed photograph of the highland rainforests of the Atherton Tableland which is home to the Fontainea picrosperma tree.
Reference(s)
- Breakthrough in the production of an acclaimed cancer-treating drug achieved by Stanford researchers (Stanford university press release, 3 October 2022).
- Cullen, J.K., Boyle, G.M., Yap, P.Y., Elmlinger, S., Simmons, J.L., Broit, N., Johns, J., Ferguson, B., Maslovskaya, L.A., Savchenko, A.I., Mirzayans, P.M., Porzelle, A., Bernhardt, P.V., Gordon, V.A., Reddell, P.W., Pagani, A., Appendino, G., Parsons, P.G. and Williams, C.M. (2021) Activation of PKC supports the anticancer activity of tigilanol tiglate and related epoxytiglianes. Sci Rep., 11(1), 207.
- Boyle, G.M., D'Souza, M.M., Pierce, C.J., Adams, R.A., Cantor, A.S., Johns, J.P., Maslovskaya, L., Gordon, V.A., Reddell, P.W. and Parsons, P.G. (2014) Intra-lesional injection of the novel PKC activator EBC-46 rapidly ablates tumors in mouse models. PLoS One, 9(10), e108887.
- QBiotics receives first registration for tigilanol tiglate with European Medicines Agency approval of STELFONTA® (QBiotics press release, 20 January 2020).
- FDA approves first intratumoral injection to treat non-metastatic mast cell tumors in dogs (FDA press release, 16 November 2020).
- De Ridder, T.R., Campbell, J.E., Burke-Schwarz, C., Clegg, D., Elliot, E.L., Geller, S., Kozak, W., Pittenger, S.T., Pruitt, J.B., Riehl, J., White, J., Wiest, M.L., Johannes, C.M., Morton, J., Jones, P.D., Schmidt, P.F., Gordon, V. and Reddell, P. (2021) Randomized controlled clinical study evaluating the efficacy and safety of intratumoral treatment of canine mast cell tumors with tigilanol tiglate (EBC-46). J Vet Intern Med., 35(1), 415–429.
- Lamont, R.W., Conroy, G.C., Reddell, P. and Ogbourne, S.M. (2016) Population genetic analysis of a medicinally significant Australian rainforest tree, Fontainea picrosperma C.T. White (Euphorbiaceae): biogeographic patterns and implications for species domestication and plantation establishment. BMC Plant Biol., 16, 57.
- Wender, P.A., Gentry, Z.O., Fanelli, D.J., Luu-Nguyen, Q.H., McAteer, O.D. and Njoo, E. (2022) Practical synthesis of the therapeutic leads tigilanol tiglate and its analogues. Nat Chem., Epub ahead of print .
7th December 2022, Tigilanol tiglate
Researchers at Stanford university have figured out a sustainable and reliable way to synthesise a promising plant-derived anticancer compound. To date, the compound appeared synthetically innacessible given its complex structure and no plausible path seemed to exist for synthesising it practically in a laboratory. The compound known as tigilanol tiglate (designated EBC-46, CHEBI:194146) was initially identified by an automated drug candidate screening process by the Australian company QBiotics [1].In nature, tigilanol tiglate is found in the seeds of the pink fruit of the blushwood tree, Fontainea picrosperma. It is proposed that tigilanol tiglate induces rapid tumour ablation, in part by an isoform-selective modulation of protein kinase C (PKC) [2]. After intratumoural administration, the compound induces a localized immune response and rupture of tumour vasculature, leading to haemorrhagic necrosis, rapid tumour cell death and subsequent clearance of the solid tumour [3]. In 2020, the European Medicines Agency and the Food and Drug Administration approved a tigilanol tiglate-based medication, sold under the brand name Stelfonta for the treatment of non-metastatic mast cell tumours in canines [4,5]. A recent clinical study showed a 75% cure rate in canines after a single intratumoural injection and an 88% remission after a second dose. Clinical trials have since commenced in human patients for the treatment of a broad range of cancers including head and neck, melanoma, and soft tissue cancers [6].
The only source of tigilanol tiglate is a tree found in a small rainforest region of Northeastern Australia. The tree's limited numbers and restricted distribution prompted consideration of designed tree plantations to address supply needs. However, this presents further challenges, for example the trees require pollination, meaning the right sort of pollinating animals must be present, and the trees are affected by seasonal and climate variations [7]. Given the immediate clinical and research value of tigilanol tiglate, Wender and colleagues at Stanford university proposed a semisynthetic route from a more readily available precursor [8].
Their research work published in the journal Nature Chemistry proposed phorbol, a plant derived compound as starting point for synthesising tigilanol tiglate [8]. Globally, around 7,000 plant species produce phorbol derivatives and phorbol-rich seeds are commercially inexpensive. The team extracted phorbol-rich oil from the seeds of Croton tiglium, a herb used in traditional Chinese medicine. After processing the oil to yield phorbol, they then had to overcome the previously insurmountable challenge of bedecking a part of the molecule, called the B ring, with carefully placed oxygen atoms that were critical for PKC activity. The team succeeded in adding extra oxygen atoms to phorbol's B ring via an ene reaction conducted under flow conditions. The team then introduced other functional groups in a stepwise, controlled manner to obtain the required spatial arrangements of the atoms. In total, 12 steps were required to produce tigilanol tiglate itself and 4-6 steps to obtain derivatives of tigilanol tiglate. It is hoped that the broader availability of tigilanol tiglate and its derivatives afforded by this approach will accelerate research into potentially new treatments [8].
The background image is a detail from a Creative Commons licensed photograph of the highland rainforests of the Atherton Tableland which is home to the Fontainea picrosperma tree.
Reference(s)
- Breakthrough in the production of an acclaimed cancer-treating drug achieved by Stanford researchers (Stanford university press release, 3 October 2022).
- Cullen, J.K., Boyle, G.M., Yap, P.Y., Elmlinger, S., Simmons, J.L., Broit, N., Johns, J., Ferguson, B., Maslovskaya, L.A., Savchenko, A.I., Mirzayans, P.M., Porzelle, A., Bernhardt, P.V., Gordon, V.A., Reddell, P.W., Pagani, A., Appendino, G., Parsons, P.G. and Williams, C.M. (2021) Activation of PKC supports the anticancer activity of tigilanol tiglate and related epoxytiglianes. Sci Rep., 11(1), 207.
- Boyle, G.M., D'Souza, M.M., Pierce, C.J., Adams, R.A., Cantor, A.S., Johns, J.P., Maslovskaya, L., Gordon, V.A., Reddell, P.W. and Parsons, P.G. (2014) Intra-lesional injection of the novel PKC activator EBC-46 rapidly ablates tumors in mouse models. PLoS One, 9(10), e108887.
- QBiotics receives first registration for tigilanol tiglate with European Medicines Agency approval of STELFONTA® (QBiotics press release, 20 January 2020).
- FDA approves first intratumoral injection to treat non-metastatic mast cell tumors in dogs (FDA press release, 16 November 2020).
- De Ridder, T.R., Campbell, J.E., Burke-Schwarz, C., Clegg, D., Elliot, E.L., Geller, S., Kozak, W., Pittenger, S.T., Pruitt, J.B., Riehl, J., White, J., Wiest, M.L., Johannes, C.M., Morton, J., Jones, P.D., Schmidt, P.F., Gordon, V. and Reddell, P. (2021) Randomized controlled clinical study evaluating the efficacy and safety of intratumoral treatment of canine mast cell tumors with tigilanol tiglate (EBC-46). J Vet Intern Med., 35(1), 415–429.
- Lamont, R.W., Conroy, G.C., Reddell, P. and Ogbourne, S.M. (2016) Population genetic analysis of a medicinally significant Australian rainforest tree, Fontainea picrosperma C.T. White (Euphorbiaceae): biogeographic patterns and implications for species domestication and plantation establishment. BMC Plant Biol., 16, 57.
- Wender, P.A., Gentry, Z.O., Fanelli, D.J., Luu-Nguyen, Q.H., McAteer, O.D. and Njoo, E. (2022) Practical synthesis of the therapeutic leads tigilanol tiglate and its analogues. Nat Chem., Epub ahead of print .

???en_GB.entity.month.date.215???, ???en_GB.entity.month.title.215???
???en_GB.entity.month.summary.215??? ???en_GB.entity.month.content.215???Reference(s)
-
???en_GB.entity.month.references.215???

???en_GB.entity.month.date.214???, ???en_GB.entity.month.title.214???
???en_GB.entity.month.summary.214??? ???en_GB.entity.month.content.214???Reference(s)
-
???en_GB.entity.month.references.214???
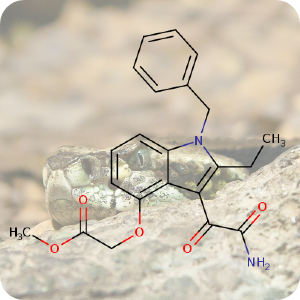
12th September 2022, Varespladib methyl
An estimated 2.7 million people are bitten by venomous snakes each year according to the World Health Organization (WHO). Each year venomous snakes kill between 81,000-138,000 people and three times that number are left with amputations and other permanent disabilities. Time is a critical factor in saving the lives of those bitten by snakes since many of the victims are located in remote, rural areas with very limited access to antivenom [1].At present, the victims of snakebite need antivenom specific to the snake species, and the medicine needs to be administered intravenously at a hospital. This presents significant logistical challenges. A new drug currently undergoing clinical trials aims to change this. Varespladib methyl (CHEBI:192805) targets secreted phospholipase A2 (sPLA2), a major component of snake venoms. The drug, manufactured by Ophirex Inc., comes in the form of a pill and can be administered in situ, and is capable of curing a broad spectrum of bites from different snake species [2]. In mouse models, the drug was effective in inhibiting neurotoxicity, abrogated lethality, and reversed neuromuscular paralysis, both immediately after envenoming and after onset of symptoms [3].
Eight clinical trial centres in the U.S. and eight in India are evaluating the effectiveness of the medicine and will provide coverage of a broad spectrum of venomous snake genera including pit vipers, elapids, and potentially exotics such as colubrids [4]. Although it will take a long time to deliver conclusive results, researchers are hopeful that their new pill could reduce the amount of intravenous antivenom that needs to be administered, reducing the need for painkillers, and shorter hospital stays for victims, thereby also lowering the overall cost of treating snake bites which is currently between $76,000 and $100,000 [2].
The background image is a detail from a Creative Commons licensed photograph of a pit viper commonly found in the U.S.
Reference(s)
- Snakebite envenoming (WHO fact sheet, 17 May 2021).
- New antivenin pill could change the way snakebites are treated (Field & Stream press release, 6 July 2022).
- Xie, C., Albulescu, L.O., Still, K.B.M., Slagboom, J., Zhao, Y., Jiang, Z., Somsen, G.W., Vonk, F.J., Casewell, N.R. and Kool, J. (2020) Varespladib inhibits the phospholipase A2 and coagulopathic activities of venom components from hemotoxic snakes. Biomedicines, 8(6), E165.
- Broad-spectrum rapid antidote: varespladib oral for snakebite (BRAVO) (ClinicalTrials.gov, NCT04996264).
4th August 2022, Varespladib methyl
An estimated 2.7 million people are bitten by venomous snakes each year according to the World Health Organization (WHO). Each year venomous snakes kill between 81,000-138,000 people and three times that number are left with amputations and other permanent disabilities. Time is a critical factor in saving the lives of those bitten by snakes since many of the victims are located in remote, rural areas with very limited access to antivenom [1].At present, the victims of snakebite need antivenom specific to the snake species, and the medicine needs to be administered intravenously at a hospital. This presents significant logistical challenges. A new drug currently undergoing clinical trials aims to change this. Varespladib methyl (CHEBI:192805) targets secreted phospholipase A2 (sPLA2), a major component of snake venoms. The drug, manufactured by Ophirex Inc., comes in the form of a pill and can be administered in situ, and is capable of curing a broad spectrum of bites from different snake species [2]. In mouse models, the drug was effective in inhibiting neurotoxicity, abrogated lethality, and reversed neuromuscular paralysis, both immediately after envenoming and after onset of symptoms [3].
Eight clinical trial centres in the U.S. and eight in India are evaluating the effectiveness of the medicine and will provide coverage of a broad spectrum of venomous snake genera including pit vipers, elapids, and potentially exotics such as colubrids [4]. Although it will take a long time to deliver conclusive results, researchers are hopeful that their new pill could reduce the amount of intravenous antivenom that needs to be administered, reducing the need for painkillers, and shorter hospital stays for victims, thereby also lowering the overall cost of treating snake bites which is currently between $76,000 and $100,000 [2].
The background image is a detail from a Creative Commons licensed photograph of a pit viper commonly found in the U.S.
Reference(s)
- Snakebite envenoming (WHO fact sheet, 17 May 2021).
- New antivenin pill could change the way snakebites are treated (Field & Stream press release, 6 July 2022).
- Xie, C., Albulescu, L.O., Still, K.B.M., Slagboom, J., Zhao, Y., Jiang, Z., Somsen, G.W., Vonk, F.J., Casewell, N.R. and Kool, J. (2020) Varespladib inhibits the phospholipase A2 and coagulopathic activities of venom components from hemotoxic snakes. Biomedicines, 8(6), E165.
- Broad-spectrum rapid antidote: varespladib oral for snakebite (BRAVO) (ClinicalTrials.gov, NCT04996264).
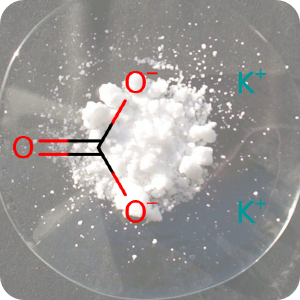
14th June 2022, Potassium carbonate
A team of researchers from Eindhoven University of Technology, TNO and its spin-out company Cellcius have invented a 'heat battery' based on salt and water that can potentially make millions of homes in Europe gas-free in the near future [1]. It took 12 years to design and develop a battery that could be transformed into a scalable solution and comes at a critical time when the EU is looking to wean itself off Russian gas dependence following the invasion of Ukraine [2]. The battery contains no toxic chemicals, rare or precious metals, and the salt contained within it is potassium carbonate (CHEBI:131526) which is harmless, cheap and abundantly available [1].The heat battery is based on an old thermochemical principle, which is that when water is added to potassium carbonate salt crystals, they absorb water, become larger and in the process release heat. The reverse reaction is also possible, where heat is used to evaporate the water, thus storing the heat energy inside the salt. The energy supply coming from renewable sources such as wind and solar tend to fluctuate significantly and require additional sources of energy to supplement it. However, storing heat within dry salt makes this new battery completely loss-free, thereby providing a very efficient way to store energy for future use [2,3].
The team have already developed a working prototype and their compact battery system is ready for real-world tests and potentially be a game-changer for energy transition. A pilot experiment is being set up later this year to test the technology in homes in the Netherlands, France, and Poland [3].
The background image is a detail from a Creative Commons licensed photograph of anhydrous potassium carbonate sample on a watch glass.
Reference(s)
- Heat batteries full of salt could 'take millions of homes off gas' (Cellcius press release, 25 April 2022).
- 'Heat battery' invention could make millions of homes gas-free (The Independent press release, 30 April 2022).
- Solving Europe's energy crisis with salt! This Eindhoven heat battery could be the key to gas-free homes (Silicon Canals press release, 27 April 2022).
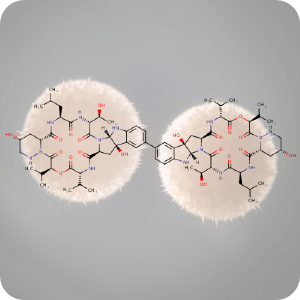
1st April 2022, Himastatin
Himastatin (CHEBI:190009), a natural product produced by the soil bacterium, Streptomyces himastatinicus, was first discovered in the 1990s. The compound is known for its antibiotic activity and when tested in animals, it was also found to have potent anticancer activity [1]. The compound is a dimer that consists of two identical subunits (monomers) that are joined together by a bond that connects a six-carbon ring in one of the monomers to the identical ring in the other monomer thus forming a carbon-carbon bond that is critical for the compound's antimicrobial activity. In previous efforts to synthesize the compound, chemists tried to make the carbon-carbon bond first, using two simple subunits, and then added more complex functional groups onto the monomers [2].However, bacteria that produce himastatin have an enzyme that can join the two monomers at the very last step of the synthesis, by turning each of the carbon atoms that need to be joined together into highly reactive radicals. Inspired by the way this reaction is performed in bacteria, chemists at MIT developed a synthetic route towards the total synthesis of this complex molecule that mimics this process [3]. Their work published in the journal Science, first synthesised complex monomers from amino acid building blocks, helped by rapid peptide synthesis technology previously developed by Pentelute's lab. The team then connected the two complex monomers together using a new dimerization strategy developed in the Movassaghi lab which is based on the oxidation of aniline to form carbon radicals in each molecule. These radicals then react to form the carbon-carbon bond that joins the two monomers together. Using this approach, dimers that contain different types of subunits, in addition to naturally occurring himastatin dimers were synthesised quickly [2,3].
One of the derivatives that the team produced had a fluorescent tag attached, which they used to visualize how the compound interacts with bacterial cells. They found that the drug accumulates in the bacterial cell membranes which led them to hypothesize that the compound kills bacteria by disrupting their cell membrane in a similar way to the FDA-approved antibiotic, daptomycin. The research team now plans to design and synthesise more structural derivatives in the hope that they may identify compounds with more potent antibiotic activity having already identified positions where they can derivatize to either enhance or retain the activity [2,3].
The background image is a detail from a Creative Commons licensed image of Streptomyces himastatinicus colonies.
Reference(s)
- Mamber, S.W., Brookshire, K.W., Dean, B.J., Firestone, R.A., Leet, J.E., Matson, J.A. and Forenza, S. (1994) Inhibition of antibacterial activity of himastatin, a new antitumor antibiotic from Streptomyces hygroscopicus, by fatty acid sodium salts. Antimicrob Agents Chemother., 38(11), 2633–2642.
- Chemical synthesis yields potential antibiotic (MIT news press release, 24 February 2022).
- D'Angelo, K.A., Schissel, C.K., Pentelute, B.L. and Movassaghi, M. (2022) Total synthesis of himastatin. Science, 375(6583), 894–899.
1st March 2022, Himastatin
Himastatin (CHEBI:190009), a natural product produced by the soil bacterium, Streptomyces himastatinicus, was first discovered in the 1990s. The compound is known for its antibiotic activity and when tested in animals, it was also found to have potent anticancer activity [1]. The compound is a dimer that consists of two identical subunits (monomers) that are joined together by a bond that connects a six-carbon ring in one of the monomers to the identical ring in the other monomer thus forming a carbon-carbon bond that is critical for the compound's antimicrobial activity. In previous efforts to synthesize the compound, chemists tried to make the carbon-carbon bond first, using two simple subunits, and then added more complex functional groups onto the monomers [2].However, bacteria that produce himastatin have an enzyme that can join the two monomers at the very last step of the synthesis, by turning each of the carbon atoms that need to be joined together into highly reactive radicals. Inspired by the way this reaction is performed in bacteria, chemists at MIT developed a synthetic route towards the total synthesis of this complex molecule that mimics this process [3]. Their work published in the journal Science, first synthesised complex monomers from amino acid building blocks, helped by rapid peptide synthesis technology previously developed by Pentelute's lab. The team then connected the two complex monomers together using a new dimerization strategy developed in the Movassaghi lab which is based on the oxidation of aniline to form carbon radicals in each molecule. These radicals then react to form the carbon-carbon bond that joins the two monomers together. Using this approach, dimers that contain different types of subunits, in addition to naturally occurring himastatin dimers were synthesised quickly [2,3].
One of the derivatives that the team produced had a fluorescent tag attached, which they used to visualize how the compound interacts with bacterial cells. They found that the drug accumulates in the bacterial cell membranes which led them to hypothesize that the compound kills bacteria by disrupting their cell membrane in a similar way to the FDA-approved antibiotic, daptomycin. The research team now plans to design and synthesise more structural derivatives in the hope that they may identify compounds with more potent antibiotic activity having already identified positions where they can derivatize to either enhance or retain the activity [2,3].
The background image is a detail from a Creative Commons licensed image of Streptomyces himastatinicus colonies.
Reference(s)
- Mamber, S.W., Brookshire, K.W., Dean, B.J., Firestone, R.A., Leet, J.E., Matson, J.A. and Forenza, S. (1994) Inhibition of antibacterial activity of himastatin, a new antitumor antibiotic from Streptomyces hygroscopicus, by fatty acid sodium salts. Antimicrob Agents Chemother., 38(11), 2633–2642.
- Chemical synthesis yields potential antibiotic (MIT news press release, 24 February 2022).
- D'Angelo, K.A., Schissel, C.K., Pentelute, B.L. and Movassaghi, M. (2022) Total synthesis of himastatin. Science, 375(6583), 894–899.

1st February 2022, 1,4-phenylenediamine
1,4-phenylenediamine (CHEBI:51403) — more commonly known as PPD is the main ingredient in permanent hair dye formulations. When applied to hair, it undergoes oxidation and turns hair into a dark colour which doesn't fade over time. In the USA, one in every three women use PPD containing hair dyes and the chemical is present in more than 1000 hair dye formulations marketed across the globe [1]. Despite its efficacy in colouring hair, PPD has a reputation for negative side effects. It can cause reactions ranging from mild skin irritation to more severe allergic contact dermatitis. Some sensitive individuals may also experience skin inflammation and eczema and in severe cases, there may also be marked reddening and swelling on the scalp and face. Alternatives dyes have been proposed but are generally not very water-soluble and the safety of some of the dyes is not well understood [2].Venkatesan and colleagues at the National University of Singapore have recently developed non-toxic hair dyes for PPD-allergic individuals [3]. The team whose work published in the journal ACS Sustainable Chemistry & Engineering synthesised seven dyes based on PPD with modifications to the aromatic amine core. These modifications were chosen to potentially make the compounds more water soluble via the addition of hydrophilic functional groups and less reactive towards skin proteins to prevent triggering an immune response and cytotoxicity. All seven compounds permanently coloured hair samples, producing a range of shades from pinks to deep blacks that did not fade, even after three weeks of daily washing. The team then examined the compounds to determine if they are skin sensitizers. Five of the modified derivatives were weak sensitizers, whereas PPD was moderate. Additional tests showed that the new compounds produced a reduced inflammatory response in cells and three of them exhibited nearly three orders of magnitude lower permeation in porcine ear skin compared to PPD. These results suggest that the new derivatives can effectively colour hair while also avoiding the potential allergenic and sensitization risks of PPD [2,3].
The background image is a detail from a Creative Commons licensed photograph of a hairdresser colouring a client's hair.
Reference(s)
- Abd-ElZaher, M.A., Fawzy, I.A., Ahmed, H.M., Abd-Allah, A.M. and Gayyed, M.F. (2012) Some toxicological health hazards associated with subchronic dermal exposure to paraphenylene-diamine (PPD): An experimental study. Egypt. J. Forensic Sci., 2, 105–111.
- New hair dyes avoid allergic reactions (ScienceDaily press release, 26 January 2022).
- Venkatesan, G., Dancik, Y., Neupane, Y.R., Karkhanis, A.V., Bigliardi, P. and Pastorin, G. (2022) Synthesis and assessment of non-allergenic aromatic amine hair dyes as efficient alternatives to paraphenylenediamine. ACS Sustainable Chem. Eng., 10(2), 838–849.
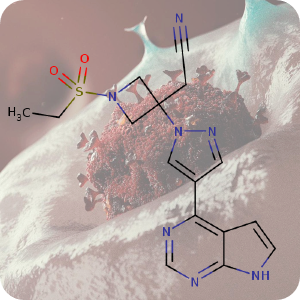
1st January 2022, Baricitinib
In January 2020, shortly after the first news of SARS-CoV-2 virus emerged from Wuhan, China and two months before the World Health Organization (WHO) declared it a global pandemic — Professor Justin Stebbing (a leading cancer expert) and his team, at Imperial College London, began working on a new therapy for COVID-19 [1].By using an artificial intelligence platform, they searched for already approved drugs that could inhibit viral entry into cells and reduce the cytokine storm — a severe and deadly immune response to the virus. Baricitinib (CHEBI:95341), an oral Janus kinase (JAK) inhibitor approved to treat rheumatoid arthritis and marketed under the brand name Olumiant™ was identified as the most promising treatment from a list of thousands of potential drugs. During the search process, the team discovered baricitinib's previously unknown anti-viral properties, in addition to its anti-inflammatory properties [2,3].
In February 2020, they published their first paper in The Lancet proposing baricitinib as a COVID-19 treatment. To date, the article has been cited over 1,000 times [2]. In June 2020, the pharmaceutical company Eli Lilly initiated a large-scale randomised COV-BARRIER phase 3 trial involving 1,525 hospitalized COVID-19 patients demonstrating baricitinib reduced mortality by 38% when combined with standard of care (E.g., corticosteroids and remdesivir) [4]. In November 2020, the FDA granted emergency use authorisation for baricitinib in combination with remdesivir to treat COVID-19 in hospitalized patients requiring supplemental oxygen or invasive mechanical ventilation [5]. In April 2021, Japan announced it had approved baricitinib to treat COVID-19 [6].
It generally takes up to 12 years for a drug to receive regulatory approval for use in patients, even if the drug has already been approved for use in other conditions after having undergone safety studies. In these unprecedented times, baricitinib received regulatory approval in record time (~10 months) [1].
The background image is a detail from a Creative Commons licensed illustration of SARS-CoV-2 virus.
Reference(s)
- How British covid breakthrough drug was built in record time - revealed (Daily Express press release, 13 November 2021).
- Richardson, P., Griffin, I., Tucker, C., Smith, D., Oechsle, O., Phelan, A., Rawling, M., Savory, E. and Stebbing, J. (2020) Baricitinib as potential treatment for 2019-nCoV acute respiratory disease. Lancet, 395(10223), e30–e31.
- Stebbing, J., Phelan, A., Griffin, I., Tucker, C., Oechsle, O., Smith, D. and Richardson, P. (2020) COVID-19: combining antiviral and anti-inflammatory treatments. Lancet Infect Dis., 20(4), 400–402.
- Marconi, V.C., Ramanan, A.V., de Bono, S., Kartman, C.E., Krishnan, V., Liao, R., Piruzeli, M.L.B., Goldman, J.D., Alatorre-Alexander, J., de Cassia Pellegrini, R., Estrada, V., Som, M., Cardoso, A., Chakladar, S., Crowe, B., Reis, P., Zhang, X., Adams, D.H. and Ely, E.W. (2021) COV-BARRIER study group. Efficacy and safety of baricitinib for the treatment of hospitalised adults with COVID-19 (COV-BARRIER): a randomised, double-blind, parallel-group, placebo-controlled phase 3 trial. Lancet Respir Med., 9(12), 1407–1418.
- Lilly and Incyte announce results from the Phase 3 COV-BARRIER study of baricitinib in hospitalized COVID-19 patients (Eli Lilly press release, 8 April 2021).
- Japan approves third drug for COVID-19 treatment (The Japan Times press release, 24 April 2021).
1st December 2021, Baricitinib
In January 2020, shortly after the first news of SARS-CoV-2 virus emerged from Wuhan, China and two months before the World Health Organization (WHO) declared it a global pandemic — Professor Justin Stebbing (a leading cancer expert) and his team, at Imperial College London, began working on a new therapy for COVID-19 [1].By using an artificial intelligence platform, they searched for already approved drugs that could inhibit viral entry into cells and reduce the cytokine storm — a severe and deadly immune response to the virus. Baricitinib (CHEBI:95341), an oral Janus kinase (JAK) inhibitor approved to treat rheumatoid arthritis and marketed under the brand name Olumiant™ was identified as the most promising treatment from a list of thousands of potential drugs. During the search process, the team discovered baricitinib's previously unknown anti-viral properties, in addition to its anti-inflammatory properties [2,3].
In February 2020, they published their first paper in The Lancet proposing baricitinib as a COVID-19 treatment. To date, the article has been cited over 1,000 times [2]. In June 2020, the pharmaceutical company Eli Lilly initiated a large-scale randomised COV-BARRIER phase 3 trial involving 1,525 hospitalized COVID-19 patients demonstrating baricitinib reduced mortality by 38% when combined with standard of care (E.g., corticosteroids and remdesivir) [4]. In November 2020, the FDA granted emergency use authorisation for baricitinib in combination with remdesivir to treat COVID-19 in hospitalized patients requiring supplemental oxygen or invasive mechanical ventilation [5]. In April 2021, Japan announced it had approved baricitinib to treat COVID-19 [6].
It generally takes up to 12 years for a drug to receive regulatory approval for use in patients, even if the drug has already been approved for use in other conditions after having undergone safety studies. In these unprecedented times, baricitinib received regulatory approval in record time (~10 months) [1].
The background image is a detail from a Creative Commons licensed illustration of SARS-CoV-2 virus.
Reference(s)
- How British covid breakthrough drug was built in record time - revealed (Daily Express press release, 13 November 2021).
- Richardson, P., Griffin, I., Tucker, C., Smith, D., Oechsle, O., Phelan, A., Rawling, M., Savory, E. and Stebbing, J. (2020) Baricitinib as potential treatment for 2019-nCoV acute respiratory disease. Lancet, 395(10223), e30–e31.
- Stebbing, J., Phelan, A., Griffin, I., Tucker, C., Oechsle, O., Smith, D. and Richardson, P. (2020) COVID-19: combining antiviral and anti-inflammatory treatments. Lancet Infect Dis., 20(4), 400–402.
- Marconi, V.C., Ramanan, A.V., de Bono, S., Kartman, C.E., Krishnan, V., Liao, R., Piruzeli, M.L.B., Goldman, J.D., Alatorre-Alexander, J., de Cassia Pellegrini, R., Estrada, V., Som, M., Cardoso, A., Chakladar, S., Crowe, B., Reis, P., Zhang, X., Adams, D.H. and Ely, E.W. (2021) COV-BARRIER study group. Efficacy and safety of baricitinib for the treatment of hospitalised adults with COVID-19 (COV-BARRIER): a randomised, double-blind, parallel-group, placebo-controlled phase 3 trial. Lancet Respir Med., 9(12), 1407–1418.
- Lilly and Incyte announce results from the Phase 3 COV-BARRIER study of baricitinib in hospitalized COVID-19 patients (Eli Lilly press release, 8 April 2021).
- Japan approves third drug for COVID-19 treatment (The Japan Times press release, 24 April 2021).

1st November 2021, Imidacloprid
Imidacloprid (CHEBI:5870), the world's leading insecticide belongs to a class of chemicals called the neonicotinoids and used in a variety of settings, from crop protection to the treatment of fleas in pets [1]. It is often used in indoor and outdoor space sprays to control the Aedes mosquito, a vector for several viruses including dengue, yellow fever, chikungunya and Zika virus. When insects land on surfaces sprayed with imidacloprid, the insecticide microcrystals are absorbed through their tarsi, subsequently binding to nicotinic acetylcholine receptors, thus disrupting their central nervous system [2].The widespread use of imidacloprid in agricultural settings has been strongly implicated in the decline of bee colonies. Given the importance of pollinators for crops and flowers, the European Union banned the use of imidacloprid in 2018 to protect bee populations, however, it is still widely used in more than 120 countries around the globe [2,3].
In an effort to reduce its environmental impact, a team of scientists from New York university have recently discovered seven new crystal polymorphs (forms) of imidacloprid, in addition to the two existing forms, one of which is used commercially. Their work published in the Journal of the American Chemical Society created new polymorphs by melting and cooling the commercial form [2].
Fast acting insecticides are necessary for rapidly killing mosquitoes before they reproduce and continue to spread diseases and are essential in reducing the likelihood of insecticide resistance. The team tested three of the new polymorphs of imidacloprid on three disease-carrying mosquitoes (Aedes, Anopheles, and Culex) in addition to fruit flies (Drosophila) [2].
All three polymorphs of imidacloprid worked significantly quicker than the commercial form, being up to nine times more potent. Generally, the more active crystal forms tend to be less stable, often transforming into their more stable counterparts, which are less active as insecticides. However, the most active crystal form of imidacloprid was easy to prepare and its microcrystals were stable to transformation in air for at least six months. It is hoped that this new polymorph could be more effective and used in smaller amounts, thereby reducing the environmental exposure and harm to non-target organisms [2].
The background image is a detail from a Creative Commons licensed photograph of a solitary bee, Anthidium florentinum, visiting a flowering plant.
Reference(s)
- Chemists discover faster-acting forms of insecticide imidacloprid (ScienceDaily press release, 12 October 2021).
- Zhu, X., Hu, C.T., Erriah, B., Vogt-Maranto, L., Yang, J., Yang, Y., Qiu, M., Fellah, N., Tuckerman, M.E., Ward, M.D. and Kahr, B. (2021) Imidacloprid crystal polymorphs for disease vector control and pollinator protection. J. Am. Chem. Soc., 143(41), 17144–17152.
- European Union expands ban of three neonicotinoid pesticides (Science press release, 27 April 2018).
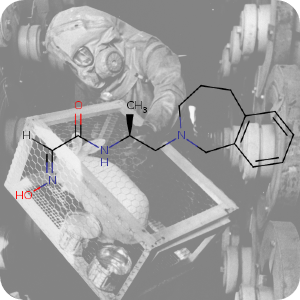
1st October 2021, LLNL-02
Nerve agents have gained increasing attention in recent times due to their use against civilian populations during the Syrian civil war in 2013, the poisoning of Sergei and Yulia Skripal in the UK in 2018 and the poisoning of Alexei Navalny in Russia in 2020, thus greatly renewing the importance of antidote development against these lethal chemicals [1,2,5].Chemical weapon nerve agents such as Sarin have the potential to cause mass casualties and act by irreversibly inhibiting the enzyme acetylcholinesterase, thus preventing the breakdown of the neurotransmitter acetylcholine. This results in profuse salivation, convulsions, and eventually death by asphyxiation [3]. The current standard treatment against their effect relies on the use of oxime compounds such as pralidoxime (2-PAM) that can effectively restore acetylcholinesterase activity. However, pralidoxime suffers from poor blood-brain barrier (BBB) penetration and therefore its efficacy is primarily limited to the peripheral nervous system (PNS), thus providing no significant protection to the central nervous system (CNS) [4,5].
To date, the blood-brain barrier has been a major obstacle to the development of effective nerve agent antidotes. Recently, a team of scientists at the Lawrence Livermore National Laboratory (LLNL) in collaboration with the United States Army Medical Research Institute of Chemical Defence (USAMRICD) have developed a new versatile antidote that is able to cross the BBB to provide protection to the brain against exposure to nerve agent poisoning. The team whose work recently published in the journal Scientific Reports, identified LLNL-02 (CHEBI:180652) using a parallel effort involving the extensive use of computational modeling, medicinal chemistry and biochemical assays [5].
LLNL-02 was able to reactivate nerve agent-adducted acetylcholinesterase. Although it was less active compared to pralidoxime, it is anticipated that its enhanced permeability into the brain would offset the decrease in activity by increasing the quantity of the bioavailable compound in the CNS. Toxicity experiments are currently ongoing, and the team plans to take the compound into the next stage of development — in vivo animal testing [5].
The background image is a detail from a Creative Commons licensed photograph of a rabbit that was used to check for leaks at a Sarin nerve gas production plant.
Reference(s)
- Chai, P.R., Hayes, B.D., Erickson, T.B. and Boyer, E.W. (2018). Novichok agents: a historical, current, and toxicological perspective. Toxicology communications, 2(1), 45–48.
- Steindl, D., Boehmerle, W., Körner, R., Praeger, D., Haug, M., Nee, J., Schreiber, A., Scheibe, F., Demin, K., Jacoby, P., Tauber, R., Hartwig, S., Endres, M. and Eckardt, K.U. (2021) Novichok nerve agent poisoning. Lancet, 397(10270), 249–252.
- Abu-Qare, A.W. and Abou-Donia, M.B. (2002) Sarin: health effects, metabolism, and methods of analysis. Food Chem Toxicol., 40(10), 1327–1333.
- Figueiredo, T.H., Apland, J.P., Braga, M. and Marini, A.M. (2018) Acute and long-term consequences of exposure to organophosphate nerve agents in humans. Epilepsia, 59(2), 92–99.
- Bennion, B.J., Malfatti, M.A., Be, N.A., Enright, H.A., Hok, S., Cadieux, C.L., Carpenter, T.S., Lao, V., Kuhn, E.A., McNerney, M.W., Lightstone, F.C., Nguyen, T.H. and Valdez, C.A. (2021) Development of a CNS-permeable reactivator for nerve agent exposure: an iterative, multi-disciplinary approach. Sci Rep., 11(1), 15567.

1st September 2021, Methanol
Natural gas consists primarily of methane that is clean, non-toxic, and has abundant natural reserves. However, methane is also a greenhouse gas whose greenhouse effect is more than 20 times than that of carbon dioxide. The conversion of methane into other value-added chemicals has been an important research area in the field of catalysis for many years. One of the most challenging processes of high industrial importance is the conversion of methane to methanol (CHEBI:17790), a simple alcohol that is liquid under ambient conditions and can be easily stored and transported compared to methane [1]. Methanol is used as an important chemical raw material to make products such as paints and plastics and as an additive to gasoline [2].Recently, a team of researchers from Stanford University and the University of Leuven has elucidated a process that could be an important step towards a methanol fuel economy with abundant methane as the feedstock. Their research work published in the journal Science utilizes a low-energy method to produce methanol from methane [3]. The process uses common crystals known as iron zeolites. When methane is infused into these porous iron zeolites, methanol is rapidly produced at room temperature with no additional requirement for heat or energy. By comparison, the conventional industrial process for making methanol from methane requires temperatures of 1000°C (1832°F) and extreme high pressure [2].
Most iron zeolites deactivate quickly and unable to process more methane, so the process peters out. The key is to get methanol out without destroying the catalyst and this is a significant barrier to scale-up at an industrial level. Scientists have therefore been keen to study ways to improve iron zeolite performance. This new study uses advanced spectroscopy to explore the physical structure of the most promising zeolites for methanol production. By choosing two iron zeolites, the team studied the physical structure of the lattices around the iron. They discovered that the reactivity varies dramatically according to the size of the pores in the surrounding crystal structure. The team refers to it as the "cage effect", as encapsulating lattice resembles a cage. When the pores in the cages are big, the active site deactivates after one reaction cycle and doesn't reactivate again. However, when the pore apertures are smaller, 40% of the active sites in zeolite are regenerated, enabling the catalytic cycle to produce methanol — a significant milestone towards an industrial-scale catalytic process [2,3].
The background image is a detail from a Creative Commons licensed photograph showing the burning of natural gas coming out of the ground in Taiwan.
Reference(s)
- Tian, Y., Piao, L. and Chen, X. (2021) Research progress on the photocatalytic activation of methane to methanol. Green Chem., 23, 3526–3541.
- International team of scientists turns methane into methanol at room temperature (Stanford University press release, 15 July 2021).
- Snyder, B.E.R., Bols, M.L., Rhoda, H.M., Plessers, D., Schoonheydt, R.A., Sels, B.F. and Solomon, E.I. (2021) Cage effects control the mechanism of methane hydroxylation in zeolites. Science, 373(6552), 327–331.
1st August 2021, Methanol
Natural gas consists primarily of methane that is clean, non-toxic, and has abundant natural reserves. However, methane is also a greenhouse gas whose greenhouse effect is more than 20 times than that of carbon dioxide. The conversion of methane into other value-added chemicals has been an important research area in the field of catalysis for many years. One of the most challenging processes of high industrial importance is the conversion of methane to methanol (CHEBI:17790), a simple alcohol that is liquid under ambient conditions and can be easily stored and transported compared to methane [1]. Methanol is used as an important chemical raw material to make products such as paints and plastics and as an additive to gasoline [2].Recently, a team of researchers from Stanford University and the University of Leuven has elucidated a process that could be an important step towards a methanol fuel economy with abundant methane as the feedstock. Their research work published in the journal Science utilizes a low-energy method to produce methanol from methane [3]. The process uses common crystals known as iron zeolites. When methane is infused into these porous iron zeolites, methanol is rapidly produced at room temperature with no additional requirement for heat or energy. By comparison, the conventional industrial process for making methanol from methane requires temperatures of 1000°C (1832°F) and extreme high pressure [2].
Most iron zeolites deactivate quickly and unable to process more methane, so the process peters out. The key is to get methanol out without destroying the catalyst and this is a significant barrier to scale-up at an industrial level. Scientists have therefore been keen to study ways to improve iron zeolite performance. This new study uses advanced spectroscopy to explore the physical structure of the most promising zeolites for methanol production. By choosing two iron zeolites, the team studied the physical structure of the lattices around the iron. They discovered that the reactivity varies dramatically according to the size of the pores in the surrounding crystal structure. The team refers to it as the "cage effect", as encapsulating lattice resembles a cage. When the pores in the cages are big, the active site deactivates after one reaction cycle and doesn't reactivate again. However, when the pore apertures are smaller, 40% of the active sites in zeolite are regenerated, enabling the catalytic cycle to produce methanol — a significant milestone towards an industrial-scale catalytic process [2,3].
The background image is a detail from a Creative Commons licensed photograph showing the burning of natural gas coming out of the ground in Taiwan.
Reference(s)
- Tian, Y., Piao, L. and Chen, X. (2021) Research progress on the photocatalytic activation of methane to methanol. Green Chem., 23, 3526–3541.
- International team of scientists turns methane into methanol at room temperature (Stanford University press release, 15 July 2021).
- Snyder, B.E.R., Bols, M.L., Rhoda, H.M., Plessers, D., Schoonheydt, R.A., Sels, B.F. and Solomon, E.I. (2021) Cage effects control the mechanism of methane hydroxylation in zeolites. Science, 373(6552), 327–331.
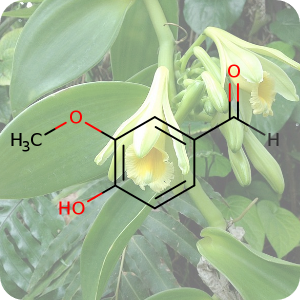
1st July 2021, Vanillin
Plastic pollution is a major environmental burden, especially in the oceans where plastics remain in the marine ecosystem for long periods due to low rates of degradation [1]. It is estimated that 311 million tons of plastics are produced annually worldwide, but only a small fraction (14%) is recycled. A significant amount of these plastics is used for the manufacture of food packaging and drinking bottles [2].A potential solution to tackling the global plastic pollution problem is to produce degradable plastics from renewable resources. Although this approach provides hope for the future, it does not help get rid of the plastics that are already in the environment [2]. Single use plastics lose about 95% of their value after use, resulting in an estimated $80-120 billion loss to the global economy each year [3]. Encouraging better collection and upcycling plastic bottles into more useful materials would have significant economic as well as environmental impact [4].
Plastic bottles are made from a polymer called poly(ethylene terephthalate) (PET). In 2016, a team of Japanese researchers from the Kyoto Institute of Technology discovered a new species of bacteria that produces enzymes which can break down PET into two environmentally benign monomers, terephthalic acid and ethylene glycol [5]. In 2020, building on this finding, a group of researchers at the University of Portsmouth and four US institutions created a super enzyme which was capable of degrading PET several times faster [6]. Researchers at the University of Edinburgh have now used bacteria to convert plastic waste into vanillin (CHEBI:18346), a widely used flavouring agent that is used extensively by food, cosmetics and pharmaceutical industries and whose global demand stood at 37,000 tonnes in 2018 and continues to grow, far exceeding the supply from natural vanilla beans [7].
The research work published in the journal Green Chemistry, used genetically engineered E-coli bacteria to transform terephthalic acid into vanillin. The scientists warmed a microbial broth containing terephthalic acid to 37°C for a day, the same conditions as for brewing beer. This converted 79% of the terephthalic acid into vanillin. The researchers will now tweak the bacteria to increase the conversion rate further and will work on scaling up the process. This work is an excellent demonstration of green chemistry where waste plastics that are harmful to the environment are turned into an important commodity with broad applications using microbes [4,7].
The background image is a detail from a Creative Commons licensed photograph of a Vanilla planifolia orchid whose pods are the source of natural vanilla.
Reference(s)
- Alabi, O.A., Ologbonjaye, K.I., Awosolu, O. and Alalade, O.E. (2019) Public and environmental health effects of plastic wastes disposal: a review. J. Toxicol. Risk Assess., 5, 021.
- Bornscheuer, U.T. (2016) Feeding on plastic. Science, 351(6278), 1154–1155.
- Walker, T.R., McGuinty, E., Charlebois, S. and Music, J. (2021). Single-use plastic packaging in the Canadian food industry: consumer behavior and perceptions. Humanit. Soc. Sci. Commun., 8, 80.
- Scientists convert used plastic bottles into vanilla flavouring (The Guardian press release, 15 June 2021).
- Yoshida, S., Hiraga, K., Takehana, T., Taniguchi, I., Yamaji, H., Maeda, Y., Toyohara, K., Miyamoto, K., Kimura, Y. and Oda, K. (2016) A bacterium that degrades and assimilates poly(ethylene terephthalate). Science, 351(6278), 1196–1199.
- Knott, B.C., Erickson, E., Allen, M.D., Gado, J.E., Graham, R., Kearns, F.L., Pardo, I., Topuzlu, E., Anderson, J.J., Austin, H.P., Dominick, G., Johnson, C.W., Rorrer, N.A., Szostkiewicz, C.J., Copié, V., Payne, C.M., Woodcock, H.L., Donohoe, B.S., Beckham, G.T. and McGeehan, J.E. (2020) Characterization and engineering of a two-enzyme system for plastics depolymerization. Proc. Natl. Acad. Sci. USA., 117(41), 25476–25485.
- Sadler, J.C and Wallace, S. (2021). Microbial synthesis of vanillin from waste poly(ethylene terephthalate). Green Chem., Advance article.

1st June 2021, Fluoren-9-one
Batteries hold the key to transitioning away from fossil fuel dependence and scientists are continuing to make great strides in producing better batteries which store more energy at lower costs, and which last longer. These results have translated into a more resilient electrical grid, increased use of electric vehicles, long-lasting laptop batteries, and greater use of energy from renewable energy sources such as solar, wind and hydro power [1].However, the most widely used grid-scale batteries use lithium-ion technology which is difficult to customize and there are also safety concerns. Thus, identifying the right materials and combining them to create a new formula for energy storage is critical to harness and store renewable energy. Redox flow batteries are a growing alternative but most use vanadium, a chemical which is expensive, prone to price fluctuations and not easily available. These traits pose barriers to widespread grid-scale energy storage. Alternative materials for flow batteries include organic molecules that are far more readily available, environmentally friendly and less expensive than vanadium. However, such molecules haven't held up well to the demands of flow-battery technology, usually petering out faster than required. There is a need for long-term stability of the molecules so that they maintain their ability to perform chemical reactions for many years [1,2].
A recent article published in the journal Science has identified fluoren-9-one (CHEBI:17922) as a potential candidate when it comes to energy storage [3]. The chemical is a bright fluorescent yellow powder that is inexpensive, readily available as a waste product from coal tar, and widely used in LED's, solar panels, pharmaceuticals, and candles [1]. The research team at the US Department of Energy's Pacific Northwest National Laboratory wanted to create an aqueous flow battery with fluoren-9-one at its heart. In order to prevent its fast degradation, the fluoren-9-one was modified into a redox reversible, water-soluble compound. Prior to this technical improvement, this chemical was not water-soluble enough and could not provide redox reversibility in aqueous solutions. The solubility of the compound was crucial in redox flow batteries and the researchers were able to increase its water solubility from almost 0 to 1.5 moles per litre, depending on the modification to the compound. The flow battery had a size of 10 cm2, about the size of a large postage stamp, and a power output of 500 milliwatts, not even enough to power a smartphone camera. But the tiny structure has tremendous potential since its energy density was more than twice that of the vanadium batteries in use today and its chemical components are long lasting, inexpensive, and widely available. In laboratory experiments, the battery operated continuously for 120 days, ending only when other equipment unrelated to the battery itself wore out. The battery went through 1,111 charging cycles — the equivalent of several years of operation under normal circumstances through which it lost less than 3 percent of its energy capacity, demonstrating a far superior lifespan to other organic flow batteries [3]. The researchers have filed a patent for their new battery design.
The background image is a detail from a Creative Commons licensed photograph of a candle with a gold stand.
Reference(s)
- Compound commonly found in candles lights the way to grid-scale energy storage (Pacific Northwest National Laboratory press release, 20 May 2021).
- Redox flow battery based on fluorenone (PV magazine press release, 26 May 2021).
- Feng, R., Zhang, X., Murugesan, V., Hollas, A., Chen, Y., Shao, Y., Walter, E., Wellala, N.P.N., Yan, L., Rosso, K.M. and Wang, W. (2021) Reversible ketone hydrogenation and dehydrogenation for aqueous organic redox flow batteries. Science, 372(6544), 836–840.
1st May 2021, PF-07321332
To date, more than 34 million people have received their 1st dose of the COVID-19 vaccine in the United Kingdom, with more than 14 million people having also received their 2nd dose (either by Oxford-AstraZenenca, Pfizer-BioNTech or Moderna, all of which requires two doses for maximum protection). But tackling the COVID-19 pandemic requires not only prevention via vaccine but also targeted drug treatment for those who contract the virus [1].The continued global impact of COVID-19 and the way in which the SARS-CoV-2, the coronavirus which causes COVID-19 is mutating, means that access to several therapeutic options now and beyond the pandemic will be critical. In February, Pfizer launched a clinical trial of a new antiviral drug. Up to 60 healthy participants aged between 18 and 60 were enrolled and given the first pill specifically designed to target SARS-CoV-2 [2].
The molecule being tested is a bespoke antiviral codenamed PF-07321332 (CHEBI:170007). The compound is a reversible covalent inhibitor that reacts with a cysteine residue in the main protease (also known as 3CL protease) of SARS-CoV-2. Inhibition of the main protease prevents the virus from cleaving long protein chains into the parts it needs to replicate [3]. PF-07321332 is being administered in combination with low doses of Ritonavir, an antiviral drug used to treat HIV since it will act as a ''booster'' to increase PF-07321332 levels in the bloodstream [2]. The drug was developed from scratch during the current pandemic by a team of 210 researchers and its synthesis was scaled up in a matter of 4 months from 7 mg to more than 1 kg. In animal studies, no significant safety risks or concerns were identified, and the drug did not cause any side-effects at any of the dose levels that will be used in the clinical trial [3].
If the trial is successful, it is possible that people who test positive for coronavirus could be sent antiviral tablets to take at home. This is the ambition of the UK Antivirals Taskforce recently announced by the UK Prime Minister, whose brief is to specifically invest in such products to bolster the UK's defences against another wave of infections. The new Antivirals Taskforce is expected to mirror the success of the Vaccines Taskforce, which secured the UK's access to a range of vaccine candidates last year [4,5]. It is hoped any such new medicines could help combat any future rise in infections and limit the impact of new COVID variants, especially over the winter flu season.
The background image is a detail from a Creative Commons licensed image of a scanning electron micrograph of an apoptotic cell (green) infected with SARS-CoV-2 (yellow).
Reference(s)
- Vaccination in United Kingdom (The UK government daily report).
- Revealed: How a single pill home cure for Covid could be available this year (The Telegraph press release, 24 April 2021).
- Pfizer unveils its oral SARS-CoV-2 inhibitor (Chemical & Engineering News press release, 7 April 2021).
- COVID-19: Britons who test positive for coronavirus could be sent antiviral tablets to take at home (Sky News press release, 21 April 2021).
- Government launches COVID-19 Antivirals Taskforce to roll out innovative home treatments this autumn (Department of Health and Social Care press release, 20 April 2021).
1st April 2021, Aetokthonotoxin
Avian vacuolar myelinopathy (AVM) was first discovered in the winter of 1994 when 29 bald eagle carcasses were found near DeGray Lake, a man-made reservoir in Arkansas. Two years later, at least 70 eagles had died across several Arkansas reservoirs. By 1998, conditions continued to deteriorate as dead birds of different species including American coots, mallards and ring-necked ducks were confirmed at ten sites across six states. All of the documented cases of this emerging disease occurred on or near man-made water reservoirs with abundant aquatic vegetation [1,2].But what particularly puzzled scientists was how these birds behaved before they died. The birds were observed suddenly flying straight into stationary objects (rock walls and trees), swimming in circles, and with their wings drooped [1]. For many years, scientists from state and federal agencies tried to figure out what was killing the birds and testing of sediment and dead birds at affected water reservoirs for xenobiotic compounds that cause AVM revealed nothing [3]. Necropsies of the birds' brains showed widespread vacuolation of white matter of the central nervous system and spinal cord which explained their uncoordinated behaviour [2].
Early studies suggested that an unknown, seasonal and environmental neurotoxin could be responsible. Recently, a paper published in the journal Science has pinpointed the exact neurotoxin responsible for the death of those bald eagles in Arkansas [4]. The research describes the series of events that leads to AVM. It's a fatal three-part process that requires invasive plants, abundant bacteria, and chemicals in the environment. It begins with the invasive plant named Hydrilla verticillata, also called waterthyme, first introduced to the United States in the 1950's as an aquarium plant. The plant is home to cyanobacteria called Aetokthonos hydrillicola which colonize up to 95% of the plant leaves. The cyanobacteria aren't themselves deadly to waterbirds but when they are exposed to the chemical bromide in the environment, they produce a dangerous neurotoxin called aetokthonotoxin (CHEBI:167886) which causes AVM in birds [4].
Previous field and laboratory studies have also demonstrated that AVM in herbivorous waterbirds due to the ingestion of H. verticillata can be transferred up the food chain to birds of prey which consume the affected waterfowl. AVM thus presents an emerging threat to multiple avian species. Furthermore, feeding trials have also confirmed neuropathy and mortality in a wide range of taxa including amphibians, reptiles, and fish [5,6]. This work highlights the role of cyanobacteria as potentially dangerous toxin producers and further research into how the toxin effects human health via the consumption of fish and waterbirds from these contaminated reservoirs is urgently required [4].
The background image is a detail from a Creative Commons licensed photograph of a bald eagle preparing to fly at Kachemak Bay, Alaska.
Reference(s)
- Fischer, J.R., Lewis-Weis, L.A., Tate, C.M., Gaydos, J.K., Gerhold, R.W. and Poppenga, R.H. (2006) Avian vacuolar myelinopathy outbreaks at a southeastern reservoir. J. Wildl. Dis., 42(3), 501–510.
- Thomas, N.J., Meteyer, C.U. and Sileo, L. (1998) Epizootic vacuolar myelinopathy of the central nervous system of bald eagles (Haliaeetus leucocephalus) and American coots (Fulica americana). Vet. Pathol., 35(6), 479–487.
- Dodder, N.G., Strandberg, B., Augspurger, T. and Hites, R.A. (2003) Lipophilic organic compounds in lake sediment and American coot (Fulica americana) tissues, both affected and unaffected by avian vacuolar myelinopathy. Sci. Total Environ., 311(1-3), 81–89.
- Breinlinger, S., Phillips, T.J., Haram, B.N., Mareš, J., Martínez Yerena, J.A., Hrouzek, P., Sobotka, R., Henderson, W.M., Schmieder, P., Williams, S.M., Lauderdale, J.D., Wilde, H.D., Gerrin, W., Kust, A., Washington, J.W., Wagner, C., Geier, B., Liebeke, M., Enke, H., Niedermeyer, T.H.J. and Wilde, S.B. (2021) Hunting the eagle killer: A cyanobacterial neurotoxin causes vacuolar myelinopathy. Science, 371(6536), eaax9050.
- Mercurio, A.D., Hernandez, S.M., Maerz, J.C., Yabsley, M.J., Ellis, A.E., Coleman, A.L., Shelnutt, L.M., Fischer, J.R. and Wilde, S.B. (2014) Experimental feeding of Hydrilla verticillata colonized by stigonematales cyanobacteria induces vacuolar myelinopathy in painted turtles (Chrysemys picta). PLoS One, 9(4), e93295.
- Lewis-Weis, L.A., Gerhold, R.W. and Fischer, J.R. (2004) Attempts to reproduce vacuolar myelinopathy in domestic swine and chickens. J. Wildl. Dis., 40(3), 476–484.
1st March 2021, Semaglutide
The negative health implications of obesity and its effects on mortality rates have been brought into stronger focus by the current COVID-19 crisis [1]. A recent international clinical trial published in The New England Journal for Medicine has shown that a third (35%) of obese patients treated with semaglutide (CHEBI:167574, brand name: Ozempic), lost at least a fifth of their total bodyweight within 68-weeks [2]. The results have been described as a gamechanger in the fight against obesity and the trial proves that it's possible to achieve weight loss through drugs which was previously only possible through lifestyle modifications and weight loss surgery [2].Semaglutide, a drug approved to treat type 2 diabetes suppresses appetite by mimicking the human glucagon-like peptide-1 (GLP-1) hormone, which is released into the blood from the gut after meals and signals to the brain that a person has eaten enough. The drug induces weight loss by reducing hunger and thereby enables people to reduce their calorie intake [3].
The randomised control trial involved 1,961 adults who were either overweight or obese (average weight 105 kg / 16.5 stone, body mass index 38 kg/m2), and took place at 129 sites in 16 countries across Asia, Europe, North America, and South America [2].
Participants received either a 2.4 mg dose of semaglutide or placebo via a weekly subcutaneous injection. Overall, 94.3% of participants completed the 68-week study. Participants taking part in the trial also received individual face-to-face or phone counselling sessions from registered dietitians every four weeks to help them adhere to the reduced-calorie diet and increased physical activity. For patients who took semaglutide, the average weight loss was 15.3 kg (2.4 stone), with a reduction in BMI of -5.54, compared to the placebo group with an observed average weight loss of 2.6 kg (0.4 stone) and a reduction in BMI of -0.92 [2].
In addition to causing weight loss, participants who took semaglutide also saw reductions in risk factors for heart disease and diabetes and reported improvements in their overall quality of life. The most common side effects reported by participants in this trial were nausea and diarrhoea, which were typically transient and mild-to-moderate [2]. In December 2020, Novo Nordisk filed semaglutide for regulatory approval for weight management with the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) [4,5].
The background image is a detail from a Creative Commons licensed image from the scroll of diseases and deformities (Yamai no Soshi), depicting a woman moneylender with obesity, considered a disease of the rich.
Reference(s)
- Stefan, N., Birkenfeld, A.L. and Schulze, M.B. (2021) Global pandemics interconnected - obesity, impaired metabolic health and COVID-19. Nat Rev Endocrinol., 17(3), 135–149.
- Wilding, J.P.H., Batterham, R.L., Calanna, S., Davies, M., Van Gaal, L.F., Lingvay, I., McGowan, B.M., Rosenstock, J., Tran, M.T.D., Wadden, T.A., Wharton, S., Yokote, K., Zeuthen, N., Kushner, R.F. and STEP 1 Study Group. (2021) Once-weekly semaglutide in adults with overweight or obesity. N. Engl. J. Med.., published online ahead of print.
- Blundell, J., Finlayson, G., Axelsen, M., Flint, A., Gibbons, C., Kvist, T. and Hjerpsted, J.B. (2017) Effects of once-weekly semaglutide on appetite, energy intake, control of eating, food preference and body weight in subjects with obesity. Diabetes Obes. Metab., 19(9), 1242–1251.
- Novo Nordisk files for US FDA regulatory approval of once-weekly semaglutide 2.4 mg for weight management (Novo Nordisk press release, 4 December 2020).
- Novo Nordisk files for EU regulatory approval of once-weekly semaglutide 2.4 mg for weight management (Novo Nordisk press release, 18 December 2020).

1st February 2021, Fluvoxamine
A new study has demonstrated that the antidepressant fluvoxamine (CHEBI:5138), a selective serotonin reuptake inhibitor (SSRI) prescribed for the treatment of obsessive-compulsive disorder (OCD), could prevent people from getting seriously ill with COVID-19, and thereby prevent hospitalisations [1].Researchers from Washington University in St. Louis, U.S., shared the results of the clinical trial that examined fluvoxamine in patients with mild COVID-19. The results indicated that if fluvoxamine is given early in the course of the disease, it significantly reduces the likelihood of hospitalisation. In addition to inhibiting serotonin reuptake, fluvoxamine also binds to a protein called the sigma-1 receptor. The researchers believe that fluvoxamine's action at the sigma-1 receptor may prevent the excessive release of cytokines, small inflammatory proteins which can exacerbate inflammatory reactions and the need for ventilator support [2].
The trial recruited 152 participants with mild forms of COVID-19 who were randomly assigned to take either 100 mg of fluvoxamine or a placebo three times daily for 15 days. As this was a remote, outpatient clinical trial, there was no face-to-face contact between participants and clinicians. Instead, the study material along with the drug were delivered to the participants homes. Out of the 80 participants who received the drug, zero hit the endpoint of clinical deterioration, as compared to the six of 72 people who were given the placebo and deteriorated. These preliminary results suggest that fluvoxamine may mitigate the risk of hospitalisation and death. A larger remote outpatient clinical trial is now underway to validate these findings [2].
The background image is a detail from a Creative Commons licensed photograph showing the main entrance to NHS Nightingale Hospital, London, UK (a temporary hospital set up by NHS England for the COVID-19 pandemic).
Reference(s)
- Trial supports use of fluvoxamine in patients with mild COVID-19 (European Pharmaceutical Review press release, 7 October 2020).
- Lenze, E.J., Mattar, C., Zorumski, C.F., Stevens, A., Schweiger, J., Nicol, G.E., Miller, J.P., Yang, L., Yingling, M., Avidan, M.S. and Reiersen, A.M. (2020) Fluvoxamine vs placebo and clinical deterioration in outpatients with symptomatic COVID-19: A randomized clinical trial. JAMA, 324(22), 2292–2300.

1st January 2021, Graphene
In 2004, two physicists working at the University of Manchester, Andre Geim and Konstantin Novoselov were playing with graphite cubes (the same material used to make pencil leads) and Scotch tape. They applied tape to a piece of graphite, then ripped the tape off, pulling flakes of graphite with it. They did this repeatedly with the flaked-off graphite, separating it into thinner and thinner flakes, until they isolated graphene (CHEBI:36973) for the very first time, a one-atom-thick planar sheet of carbon atoms arranged in a honeycomb crystal lattice for which the pair were awarded the Nobel Prize in Physics in 2010 [1].Graphene's unique strength, flexibility and electrical conductivity offers tremendous potential for use in several applications and is often touted as the prime candidate to replace silicon in transistors in the future [2]. However, graphene is not a semiconducting material and currently has limited use in electronics since it lacks a band gap (the energy difference between the valence and the conduction bands in semiconductors or metals). To produce switching devices such as transistors, a band gap is required which allows the movement of electrons from the valence to the conduction bands and vice-versa, so the material can switch electrical currents on and off. In graphene, the electrons roam freely across the sheet of carbon, conducting electric charge with extremely low resistance which makes it useless as a material for use in transistors since it cannot give the transistor the ability to switch off [3].
Recently, a team led by Professor Federico Rosei at the Institut national de la recherche scientifique (INRS) have succeeded in modifying graphene so as to create a band gap. They demonstrated a novel process of modifying the structure and properties of graphene by a chemical reaction known as photocycloaddition which modifies the bonds between atoms using ultraviolet (UV) light. Their research work published in the journal Nature Chemistry could pave the way to designing and engineering graphene-based optoelectronic and microelectronic devices [4].
The background image is a detail from a Creative Commons licensed image showing the crystalline structure of graphene as a hexagonal lattice made of carbon atoms.
Reference(s)
- The graphene story: how Andrei Geim and Kostya Novoselov hit on a scientific breakthrough that changed the world... by playing with sticky tape (The Independent press release, 18 March 2013).
- Palmer, J. (2008) Graphite – not graphene – could replace silicon transistors. NewScientist, 197(2638), 24.
- Maharubin, S., Zhang, X., Zhu, F., Zhang, H-C., Zhang, G. and Zhang, Y. (2016) Synthesis and applications of semiconducting graphene. Journal of Nanomaterials, 6375962.
- Yu, M., Chen, C., Liu, Q., Mattioli, C., Sang, H., Shi, G., Huang, W., Shen, K., Li, Z., Ding, P., Guan, P., Wang, S., Sun, Y., Hu, J., Gourdon, A., Kantorovich, L., Besenbacher, F., Chen, M., Song, F. and Rosei, F. (2020) Long-range ordered and atomic-scale control of graphene hybridization by photocycloaddition. Nat Chem., 12(11), 1035–1041.
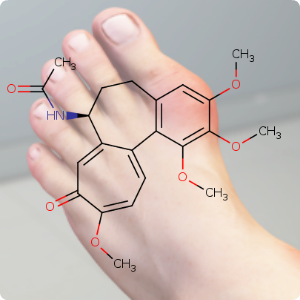
1st December 2020, Colchicine
Inflammation caused by an overactive immune response is a key element of severe COVID-19, and can lead to lung damage, the necessity for mechanical ventilation, and death [1]. In the latest effort to repurpose existing medicines to fight the COVID-19 pandemic, the anti-inflammatory drug colchicine (CHEBI:27882) is currently being investigated as a possible treatment in the randomised evaluation of COVID-19 therapy (RECOVERY) trial [2].The RECOVERY trial which is taking place across 176 hospital sites in the UK with over 18,000 patients recruited so far is considered as the the world's largest clinical trial of treatments for patients hospitalized with COVID-19 [2]. Some of the other treatments being tested in the RECOVERY trial include the immunosuppressive drug Tocilizumab and REGEN-COV2 (the Regeneron antibody cocktail which was used to treat U.S. President Donald Trump's COVID-19 symptoms) [3].
The RECOVERY trial has already shown that treatment with the steroid dexamethasone, another anti-inflammatory drug, can reduce deaths in the most severely ill patients, and therefore colchicine is considered an attractive drug to evaluate in the trial as it is very well understood, inexpensive and widely available [4]. Colchicine is derived from the bulb-like corms of the Colchicum autumnale plant and has a wide range of anti-inflammatory effects. It has been used for centuries to treat gout and, more recently, other inflammatory conditions [5].
Around 2,500 patients hospitalised with COVID-19 in the UK will be given the drug plus usual standard-of-care and compared to 2,500 patients who will receive the usual standard-of-care alone. An initial dose of 1,000 micrograms will be followed up with 500 micrograms every 12 hours for ten days. The researchers are primarily focused on seeing if the treatment reduces mortality after 28 days. Other outcomes of the trial include the impact on hospital stay and the need for ventilation. It will take several months before there is enough evidence to conclude whether colchicine has a significant benefit in COVID-19 patients [2].
The background image is a detail from a Creative Commons licensed image showing a foot affected by gout.
Reference(s)
- Ledford, H. (2020) How does COVID-19 kill? Uncertainty is hampering doctors' ability to choose treatments. Nature, 580(7803), 311–312.
- Colchicine to be investigated as a possible treatment for COVID-19 in the RECOVERY trial (RECOVERY trial press release, 27 November 2020).
- FDA authorizes Regeneron's Covid treatment, taken by Trump, for emergency use (CNBC press release, 21 November 2020).
- RECOVERY Collaborative Group, Horby, P., Lim, W.S., et al. (2020) Dexamethasone in hospitalized Patients with Covid-19 - Preliminary Report. N. Engl. J. Med., NEJMoa2021436.
- Dasgeb, B., Kornreich, D., McGuinn, K., Okon, L., Brownell, I. and Sackett, D.L. (2018) Colchicine: an ancient drug with novel applications. Br. J. Dermatol., 178(2), 350–356.
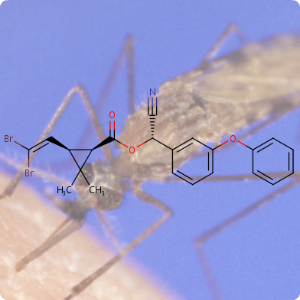
1st November 2020, Deltamethrin
Malaria, an infectious disease caused by parasites of the genus Plasmodium is a global health challenge, especially in developing countries. According to the World Health Organisation, in 2018, there were an estimated 228 million cases of malaria and 405,000 deaths occurred worldwide. The parasite is transmitted to people through the bites of infected female Anopheles mosquitoes [1].Pyrethroids are the most widely used insecticides for malaria control due to their relatively low toxicity to humans, high insect lethality and cost-effectiveness. Among pyrethroids, deltamethrin (CHEBI:4388) is commonly used for indoor residual spraying and for treating bed nets, providing malaria control for millions of people. Unfortunately, the development of insecticide resistance in mosquito vectors is a growing problem and if left unchecked could lead to a substantial increase in malaria incidence and mortality [2]. Urgent action is therefore needed to prevent the development of resistance and maintain the effectiveness of existing vector control interventions.
A team of researchers from New York university have recently demonstrated that the use of more active crystal forms of insecticides is a simple and powerful strategy for improving commercially available insecticides for malaria control, circumventing the need for developing new products in the ongoing fight against malaria. Their research work published in the journal Proceedings of the National Academy of Sciences (PNAS) showed that by heating deltamethrin to 110°C for a few minutes and leaving it to cool down to room temperature, resulted in a new crystallized form of deltamethrin which was 12 times more fast-acting compared to the commercially available form when tested on Anopheles quadrimaculatus and Aedes aegypti mosquitoes [3].
The background image is a detail from a Creative Commons licensed photograph of the Anopheles gambiae mosquito, the most efficient vector of human malaria in sub-Saharan Africa.
Reference(s)
- World malaria report 2019 (World Health Organisation, 4 December 2019).
- Mandeng, S.E., Awono-Ambene, H.P., Bigoga, J.D., Ekoko, W.E., Binyang, J, et al. (2019) Spatial and temporal development of deltamethrin resistance in malaria vectors of the Anopheles gambiae complex from North Cameroon. PLOS ONE, 14(2), e0212024.
- Yang, J., Erriah, B., Hu, C.T., Reiter, E., Zhu, X., López-Mejías, V., Carmona-Sepúlveda, I.P., Ward, M.D. and Kahr, B. (2020) A deltamethrin crystal polymorph for more effective malaria control. Proc. Natl. Acad. Sci. USA, 117(43), 26633–26638.
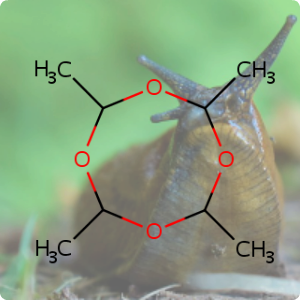
1st October 2020, Metaldehyde
Slugs cause significant damage to plants and crops, particularly cereals, potatoes, and oil seed rape and are regarded as arable farming's number one pest [1]. Metaldehyde (CHEBI:81931) is a cyclic tetramer of acetaldehyde whose crystal structure was first determined in 1936 [2]. It initially found use as a solid fuel firelighter, sold as 'Meta-Fuel' but nowadays, it is used as a molluscicide for the control of slugs and snails in agricultural fields and domestic gardens. It kills slugs and snails by disrupting their ability to produce mucus thereby reducing their digestion and mobility thus making them susceptible to dehydration [3]. However, slug pellets containing metaldehyde pose a serious risk to birds, fish and mammals (E.g. hedgehogs) which may eat the pellets, contaminated slugs or other creatures which have consumed slugs or pellets [4].The UK Department for Environment, Food and Rural Affairs (DEFRA) have recently announced that metaldehyde use will be banned in the UK from 1st April 2022. The ban was originally announced by the then environment secretary Michael Gove in December 2018 and was supposed to come into force in spring this year but was delayed when Chiltern Farm Chemicals, one of the UK's largest suppliers of molluscicides, won a High Court ruling to overturn the decision [5,6].
The latest ban will be phased over an 18-month period and gives growers plenty of time to switch to alternative measures [4]. DEFRA suggest that growers could switch to controlling slugs using pesticides containing ferric phosphate, which have been proven to be just as effective, but which don't carry the same risks to wildlife and the environment as metaldehyde. When applied to soil, ferric phosphate pellets have the ability to affect the calcium metabolism in the gut system of slugs thus causing them to stop feeding and die within three to six days [7].
The background image is a detail from a Creative Commons licensed photograph of a slug.
Reference(s)
- Slugcontrol: A grower's guide (A De Sangosse booklet).
- Pauling, L. and Carpenter, D.C. (1936) The crystal structure of metaldehyde. J. Am. Chem. Soc., 58(7), 1274–1278.
- Thomas, J.C., Helgason, T., Sinclair, C.J. and Moir, J.W.B. (2017) Isolation and characterization of metaldehyde-degrading bacteria from domestic soils. Microb. Biotechnol., 10(6), 1824–1829.
- Ban on the use of metaldehyde announced (DEFRA press release, 21 September 2020).
- Restrictions on the use of metaldehyde to protect wildlife (DEFRA press release, 19 December 2018).
- Chiltern case overturns UK metaldehyde ban (AgriTrade News press release, 2 August 2019).
- Iron (ferric) phosphate fact sheet (The United States environment protection agency).

1st September 2020, Ammonium nitrate
On 4th August 2020, a huge blast devastated the Beirut port area, killing at least 220 people, injuring more than 5,000 and leaving an estimated 300,000 people homeless. The blast was so powerful that it was heard 150 miles away in Cyprus and sent a massive orange fireball into the sky and resulted in a huge shock wave which damaged buildings, overturned cars and shook the ground across the Lebanese capital [1].The cause of the blast was ammonium nitrate (NH4NO3, CHEBI:63038), a chemical first synthesised in 1659 by the German chemist Johann Rudolf Glauber (1604–1670) by combining nitric acid and ammonium carbonate. Glauber called the substance 'nitrum flammans', Latin for 'flaming nitre', due to its tendency to explode when exposed to heat [2]. It has been reported that 2,750 tons of this chemical had been stored in a warehouse in the port for over six years without proper safety controls after it was unloaded from MV Rhosus, a cargo ship which was impounded by port officials in 2013. However, it remains unclear what caused the chemical to ignite [3].
A white crystalline solid, millions of tonnes of ammonium nitrate is produced and shipped around the world every year. One of the most widely used fertilisers, it is added to soil in granule form to provide the nitrogen that plants need to grow. It is also an important component in many types of mining explosives, where it's mixed with fuel oil and detonated by an explosive charge. Terrorist groups and militants have also made it their weapon of choice (e.g. the Oklahoma City bombing in 1995) [4]. Pure ammonium nitrate is not itself very combustible, but as a strong oxidizer, it intensifies combustion and allows other substances to ignite more readily. For these reasons, it must not be stored near combustible substances and must be kept away from sources of heat. In fact, several countries in the European Union use calcium ammonium nitrate as a fertilizer, which is much safer [5,6].
Serious industrial disasters involving ammonium nitrate are not new [7]. In 1921, an explosion at a BASF plant in Oppau, Germany, killed 561 people. More recently, in 2015, the detonation of around 800 tonnes of ammonium nitrate in the port of Tianjin, China, killed 173 people [1]. Despite its dangers, the legitimate uses of ammonium nitrate in agriculture, construction and mining have made it indispensable.
The background image is a detail from a Creative Commons licensed photograph showing the port of Beirut after the blast.
Reference(s)
- Why Beirut's ammonium nitrate blast was so devastating (Nature press release, 10 August 2020).
- Hongyu, Y., Defang, D., Hanyu, L., Ting, Y., Fubo, T., Kuo, B., Da, L., Zhonglong, Z., Bingbing, L. and Tian, C. (2016) Ab initio molecular dynamic study of solid-state transitions of ammonium nitrate. Sci. Rep., 6(18918), 1–9.
- Beirut explosion: What we know so far (BBC press release, 11 August 2020).
- Rao, P.S. (2014) Ammonium nitrate. Encyclopedia of Toxicology (3rd Edition), 209–211.
- Ammonium nitrate: what is it and why did it cause the blast in Beirut? (The Telegraph press release, 7 August 2020).
- Poplawski, D., Hoffmann, J. and Hoffmann, K. (2016) Effects of carbonate minerals on the thermal stability of fertilizers containing ammonium nitrate. J. Therm. Anal. Calorim., 124, 1561–1574.
- Babrauskas, V. (2016) Explosions of ammonium nitrate fertilizer in storage or transportation are preventable accidents. J. Hazard. Mater., 304, 134–149.

1st August 2020, Sodium hypochlorite
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the virus which causes COVID-19, is spread commonly via invisible respiratory droplets sent into the air when an infected person sneezes or coughs. The droplets are then inhaled by nearby people or contaminate surfaces which others then touch [1]. There is evidence to suggest that the virus is also present in faecal matter, so anyone not washing their hands thoroughly after visiting the toilet can potentially contaminate anything they touch [2].Coronaviruses are particularly resilient in terms of where they can survive – the virus can survive on cardboard for up to 24 hours, and up to 2-3 days on plastic and stainless-steel surfaces [3]. Disinfecting surfaces that serve as a virus reservoir is vital to prevent the spread of the virus, which as of July 2020 has infected more than 17 million people and killed more than 670,000 people worldwide [4].
In many cities across the globe, mass disinfections by workers in protective suits resembling characters from the comedy film ''Ghostbusters'' have become a common sight [5]. The most common disinfectant used is sodium hypochlorite (CHEBI:32146, the active ingredient in household bleach), demand for which has increased dramatically since the outbreak of the COVID-19 pandemic. In some cities, disinfectant tunnels have also been installed in many public places in which people walk through while a mist of sodium hypochlorite solution is sprayed on them to clear any viruses [6].
Sodium hypochlorite was first synthesised in 1789 by the French chemist Claude Louis Berthollet (1748–1822) and is an effective disinfectant against bacteria, viruses, and fungi. At first it was used as a bleaching agent to bleach cotton, and later used to remove stains from clothes and for the removal of odours. It is commonly used to purify drinking water and for keeping swimming pools sanitised [7].
However, the chemical is corrosive and toxic, can cause eye and skin irritation, nausea and vomiting, and bronchospasm due to inhalation. Its use in what are largely confidence building measures to contain the virus have been criticised by health experts as they do not help to reduce a person's ability to transmit the virus through droplets or close contact [5,8].
The background image is a detail from a Creative Commons licensed photograph of workers wearing protective gear and spraying disinfectant in Taiwan.
Reference(s)
- Does disinfecting surfaces really prevent the spread of coronavirus? (Science Magazine, 12 March 2020).
- Wang, W., Xu, Y., Gao, R., Lu, R., Han, K., Wu, G. and Tan, W. (2020) Detection of SARS-CoV-2 in different types of clinical specimens. JAMA, 323(18), 1843–1844.
- van Doremalen, N., Bushmaker, T., Morris, D.H., Holbrook, M.G., Gamble, A., Williamson, B.N., Tamin, A., Harcourt, J.L., Thornburg, N.J., Gerber, S.I., Lloyd-Smith, J.O., de Wit, E. and Munster, V.J. (2020) Aerosol and surface stability of SARS-CoV-2 as compared with SARS-CoV-1. N Engl J Med., 382(16), 1564–1567.
- Worldmeter COVID-19 Data (31 July 2020).
- Coronavirus: are Asia's mass disinfections of public spaces helpful or hazardous? (South China Morning Post press release, 1 April 2020).
- Biswal, M., Kanaujia, R., Angrup, A. and Ray, P. (2020) Disinfection tunnels: potentially counterproductive in the context of a prolonged pandemic of COVID-19. Public Health, 183, 48–49.
- Peck, B., Workeneh, B., Kadikoy, H., Patel, S.J. and Abdellatif, A. (2011) Spectrum of sodium hypochlorite toxicity in man - also a concern for nephrologists. NDT Plus, 4(4), 231–235.
- Sodium hypochlorite toxicological overview (Public Health England).
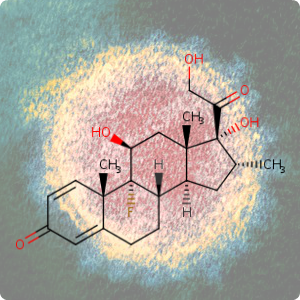
1st July 2020, Dexamethasone
Since the SARS-CoV-2 virus emerged in China late last year, there have been more than 10 million new cases of the disease worldwide, nearly half of them in the United States and Europe. To date more than 500,000 people have lost their lives globally due to the pandemic [1].In a recent clinical trial, dexamethasone (CHEBI:41879, brand name: Decadron), a cheap and widely used corticosteroid has been hailed as a major breakthrough for treating critically ill hospital patients with COVID-19 [2]. The drug, first synthesised in 1957 by the Nobel laureate Philip Showalter Hench (1896–1965) and listed on the WHO Model List of Essential Medicines, is a widely prescribed anti-inflammatory medication used to decrease swelling (edema) associated with tumours of the spine and brain, and for treating allergic reactions, arthritis, breathing problems, and certain skin and eye conditions [3].
The Randomised Evaluation of COVID-19 therapy (RECOVERY) trial which was launched in March and led by a team from Oxford University is one of the world's largest randomized controlled clinical trials for COVID-19 treatments. More than 11,500 patients have been enrolled from over 175 NHS hospitals in the UK. Its goal is to identify treatments that may be beneficial to patients hospitalised with suspected or confirmed COVID-19 and the trial is testing a range of potential treatment options, including dexamethasone [4].
The dexamethasone arm enrolled 2,104 patients who received the drug at a dose of 6 mg per day for 10 days and were compared with 4,321 patients who received the usual care for coronavirus infection. The survival benefit of dexamethasone was most remarkable among critically ill patients on ventilators. In the trial, the drug reduced deaths by one-third in ventilated patients. For patients who received oxygen therapy and were not on ventilators, the risk of dying was reduced by 20%. However, the drug had no effect on patients with less severe cases of COVID-19 (those who were not receiving oxygen or ventilation) [4].
Soon after the results were released, the UK government announced that it had immediately authorised the NHS to use dexamethasone for patients hospitalized with COVID-19 who require oxygen, including those on ventilators and that stockpiles of the drug were sufficient to treat 200,000 people [5]. It has been suggested that had the drug been used to treat patients from the onset of the pandemic, around 5000 lives could have been saved in the UK [6].
The background image is a detail from a Creative Commons licensed image of SARS-CoV-2, the virus that causes COVID-19 generated using transmission electron microscopy.
Reference(s)
- Coronavirus: Covid-19 death toll hits 500,000 worldwide (BBC press release, 28 June 2020).
- Low-cost dexamethasone reduces death by up to one third in hospitalised patients with severe respiratory complications of COVID-19 (University of Oxford press release, 16 June 2020).
- Decadron (FDA-approved drugs information).
- Horby, P., Lim, W.S., Emberson, J., Mafham, M., Bell, J., Linsell, L., Staplin, N., Brightling, C., Ustianowski, A., Elmahi, E., Prudon, B., Green, C., Felton, T., Chadwick, D., Rege, K., Fegan, C., Chappell, L.C., Faust, S.N., Jaki, T., Jeffery, K., Montgomery, A., Rowan, K., Juszczak, E., Baillie, J.K., Haynes, R., Landray, M.J. and RECOVERY Collaborative Group (2020) Effect of dexamethasone in hospitalized patients with COVID-19: preliminary report. medRxiv, in press.
- World first coronavirus treatment approved for NHS use by government (Department of health and social care press release, 16 June 2020).
- Coronavirus: dexamethasone proves first life-saving drug (BBC press release, 16 June 2020).
1st June 2020, Liquiritin
As the number of coronavirus cases increase each day, scientists worldwide are in a race to develop new treatments and a vaccine for the disease. Liquorice is a root extract of Glycyrrhiza glabra, a flowering plant that is native to southern Europe, the Middle East and Asia. It is one of the oldest used herbs in ancient medicine and its history can be traced back as far as 4000 BC. The root extract has a sweet flavour and consists of ~300 active compounds [1].The plant has a long history of medicinal uses and has been fully discussed in Ayurvedic medicine where people in ancient Hindu culture used it for improving sexual vigour. It is also mentioned in ancient Egyptian, Greek, and Roman texts for the treatment of chills, colds and coughs. The Chinese used it as a tea for strength and endurance and Alexander the Great and his army used it to quench thirst. Large quantities of it were also found in Tutankhamun's tomb among his other treasures – it was believed it could be used to prepare a sweet drink in the afterlife [2]. Today however, most of us would associate liquorice with the confectionary industry [3].
Traditional Chinese medicine (TCM) has always been a popular choice for treating a variety of conditions amongst the Chinese population. In TCM, liquorice is sold under the name 'Gan Cao' and used as a tonic to rejuvenate the heart and spleen and for treatment of several conditions including gastro and respiratory diseases, ulcers, skin disorders and cold symptoms. It is also used to harmonise herbal formulas and reduce the toxic action of other herbs [2].
Since the recent coronavirus crisis, TCM is also being promoted by some as a source of potential treatments for COVID-19, although many scientists remain unconvinced of its benefits [4]. An example of a recent study that combines compounds derived from TCM with contemporary drug discovery approaches was carried out by Chinese researchers. Their artificial intelligence approach identified liquiritin (CHEBI:80845), a compound found in liquorice extract. When tested in vitro, liquiritin prevented the replication of the SARS-CoV-2 virus in monkey cells. The research team also tested liquiritin in mice and didn't observe any toxicity or side effects. However, the study has not yet been peer-reviewed [5]. Furthermore, there are many other challenges that need to be taken into account when translating laboratory results such as these into safe and effective treatments.
The background image is a detail from a Creative Commons licensed photograph of the Glycyrrhiza glabra plant.
Reference(s)
- Deutch, M.R., Grimm, D., Wehland, M., Infanger, M. and Krüger, M. (2019) Bioactive candy: effects of licorice on the cardiovascular system. Foods, 8(495), 1–20.
- Öztürk, M., Altay, V., Hakeem, K.R. and Akçiçek, E. (2018) Liquorice: from botany to phytochemistry. Springer briefs in plant science, Springer eBook.
- Omar, H.R., Komarova, I., El-Ghonemi, M., Fathy, A., Rashad, R., Abdelmalak, H.D., Yerramadha, M.R., Ali, Y., Helal, E. and Camporesi, E.M. (2012) Licorice abuse: time to send a warning message. Ther. Adv. Endocrinol. Metab., 3(4), 125–138.
- Beijing pushes traditional Chinese medicine as coronavirus treatment despite questions over benefits (South China Morning Post press release, 23 March 2020).
- Zhu, J., Deng, Y-Q., Wang, X., Li, X-F., Zhang, N-N., Liu, Z., Zhang, B., Qin, C-F. and Xie, Z. (2020) An artificial intelligence system reveals liquiritin inhibits SARS-CoV-2 by mimicking type I interferon. bioRxiv, in press.
1st May 2020, Hydroxychloroquine
Tradition has it that in 1638 when Lady Ana de Osorio, the Countess of Chinchón, the wife of the Viceroy of Peru, fell ill with malarial fever, she was treated with the bark of a tree (much later named the Cinchona tree in her honour by the Swedish botanist Carl Linnaeus). It was at the recommendation of the Spanish Jesuit missionaries who were taught the healing power of the bark by the native Incas. Her recovery was dramatic and when she returned to Spain, she brought with her large supplies of the powdered bark, originally known as 'Jesuit's Powder' [1,2].Almost 200 years later, the active substance, quinine, was isolated from the bark by two French chemists, Pierre Joseph Pelletier (1788–1842) and Joseph Bienaimé Caventou (1795–1877). Quinine soon became an essential component in folk medicine and remained the mainstay of malaria treatment until 1920's, when more effective anti-malarials became available [2].
In 1934, Hans Andersag (1902–1955), a German scientist working at Bayer was trying to discover new antimalarial compounds and developed a compound that was named resochin but it was considered too toxic for human use. He also synthesised a less toxic derivative known as sontochin. When the Japanese took over Java during World War II, the world supply of quinine was cut off and a joint effort by American, British and Australian scientists to discover new antimalarials was initiated. Around 16,000 compounds were synthesised and tested. Resochin was one of the first compounds tested but was again considered too toxic and was dropped [2,3].
An interest in resochin was rekindled when allied soldiers captured North Africa; they obtained data and samples of German-manufactured sontochin and handed it over to the Americans. They made slight changes to the chemical structure to enhance its efficacy, and named their new drug chloroquine. When American researchers compared chloroquine to the supposedly toxic resochin, they realised they were identical. Chloroquine later proved to be the most effective antimalarial ever and widely used throughout the world [3]. In 1945, a modification to chloroquine via hydroxylation led to the development of hydroxychloroquine (CHEBI:5801), a safer alternative with fewer side-effects. Hydroxychloroquine was approved by the US Food and Drug Administration (FDA) for the treatment of lupus and rheumatoid arthritis in 1955 [4].
In search for new drugs to treat COVID-19, repositioning old drugs for use as an antiviral treatment is an attractive strategy since knowledge of the safety profile, side effects and drug interactions will be well known. In February this year, Chinese researchers demonstrated that hydroxychloroquine is effective in cell culture studies against SARS-CoV-2, the coronavirus that causes COVID-19 [5].
The hype around these promising initial results, have prompted many, including US President Donald Trump, to tout hydroxychloroquine as a game-changer in the fight against COVID-19, prompting the World Health Organisation (WHO) to add hydroxychloroquine to a list of potential therapies to investigate [6]. Media hype has led to some countries stockpiling hydroxychloroquine; India has banned all exports of the drug, resulting in shortages for patients who rely on the drug to treat their autoimmune disease [7].
Whether hydroxychloroquine works in vivo is not yet proven; a few recent clinical trials, some hastily designed and conducted in China and France have given conflicting results [8,9]. In randomised controlled trials against a number of viruses, including influenza, hydroxychloroquine doesn't work at all. One potential drawback to the use of hydroxychloroquine is the potential risk of side effects, with 'drug-induced sudden cardiac death' being previously reported from off-label use [6].
The background image is a detail from a Creative Commons licensed image of a scanning electron micrograph showing SARS-CoV-2 emerging from the surface of cells.
Reference(s)
- Achan, J., Talisuna, A.O., Erhart, A., Yeka, A., Tibenderana, J.K., Baliraine, F.N., Rosenthal, P.J. and D'Alessandro, U. (2011) Quinine, an old anti-malarial drug in a modern world: role in the treatment of malaria. Malar J., 10(144), 1–12.
- Rosenthal, P.J. (2001) Antimalarial chemotherapy: mechanisms of action, resistance, and new directions in drug discovery. Chapter 2: The history of antimalarial drugs. Humana Press, 15–25.
- Institute of Medicine (2004) Saving lives, buying time: Economics of malaria drug in an age of resistance. Part 2: Malaria basics, a brief history of malaria. The National Academies Press, 125–134.
- Shippey, E.A., Wagler, V.D. and Collamer, A.N. (2018) Hydroxychloroquine: an old drug with new relevance. Cleve Clin J Med., 85(6), 459–467.
- Liu, J., Cao, R., Xu, M., Wang, X., Zhang, H., Hu, H., Li, Y., Hu, Z., Zhong, W. and Wang, M. (2020) Hydroxychloroquine, a less toxic derivative of chloroquine, is effective in inhibiting SARS-CoV-2 infection in vitro. Cell Discovery, 6(16), 1–4.
- Owens, B. (2020) Excitement around hydroxychloroquine for treating COVID-19 causes challenges for rheumatology. The Lancet Rheumatology, in press.
- Hydroxychloroquine: India agrees to release drug after Trump retaliation threat (BBC press release, 7 April 2020).
- Chen, J., Liu, D., Liu, L., Liu, P., Xu, Q., Xia, Lu., Ling, Y., Huang, D., Song, S., Zhang, D., Qian, Z., Li, T., Shen, Y. and Lu, H. (2020) A pilot study of hydroxychloroquine in treatment of patients with common coronavirus disease-19 (COVID-19). J Zhejiang Univ (Med Sci), 49(1), 0–0.
- Gautret, P., Lagier, J-C., Parola, P., Hoang, V.T., Meddeb, L., Mailhe, M., Doudier, B., Courjon, J., Giordanengo, V., Vieira, V.E., Dupont, H.T., Honoré, S., Colson, P., Chabrière, E., Scola, B.L. Rolain, J-M., Brouqui, P. and Raoult, D. (2020) Hydroxychloroquine and azithromycin as a treatment of COVID-19: results of an open-label non-randomized clinical trial. International Journal of Antimicrobial Agents, in press.
1st April 2020, Remdesivir
Wuhan, the city in China where the coronavirus disease (COVID-19) pandemic caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) began has partially reopened after more than two months under lockdown. With new infections dwindling, shops and restaurants have reopened for business and life is slowly returning back to normal in China [1]. Meanwhile, the virus continues to spread rapidly in other countries across the globe. The US now has the highest number of confirmed infections, currently standing at 164,000. As of 30 March 2020, more than 755,000 infections have been confirmed globally, resulting in over 36,000 deaths. Since many of those with milder symptoms have not been tested and counted, the true figure for the number of people infected with coronavirus is thought to be much higher [2].In the absence of any established treatments, several trials into different drugs and experimental therapies have been initiated. Many see Gilead Sciences experimental drug, remdesivir (initially known as GS-5734, CHEBI:145994) the monophosphoramidate prodrug of the C-adenosine nucleoside analogue GS-441524 (CHEBI:147281), as a hope of an effective treatment of COVID-19. The drug, originally developed to treat Ebola and previously studied in patients in the Democratic Republic of the Congo, is considered by a World Health Organization (WHO) panel to be the most promising therapeutic candidate based on its broad antiviral spectrum [3,4].
Coronaviruses replicate by copying their genetic material using an enzyme known as the RNA-dependent RNA polymerase. Remdesivir can block replication of a variety of coronaviruses in cell cultures and animal models including SARS-CoV and MERS-CoV, but until now its mode of action has been unclear. Using polymerase enzymes from the coronavirus which causes MERS, scientists from the University of Alberta and Gilead Sciences found that remdesivir halts genome replication by mimicking an RNA building block. When remdesivir gets incorporated into viral RNA, it prevents the polymerase enzyme from adding further RNA units thereby halting viral replication [5].
In late January, remdesivir was given to a male COVID-19 patient in the US on a compassionate-use basis. The results were remarkable – almost all of his symptoms disappeared following treatment [6]. Since then, clinical testing with remdesivir has moved at an accelerated pace. Gilead Sciences has recently initiated two phase III trials in adults diagnosed with COVID-19. Both trials will test remdesivir's effectiveness in reducing patient symptoms following treatment with varying doses delivered intravenously, while closely monitoring for any adverse side effects [7].
The background image is a detail from a Creative Commons licensed illustration of SARS-CoV-2 virus.
Reference(s)
- Coronavirus cradle Wuhan partly reopens after lockdown (BBC press release, 28 March 2020).
- Coronavirus: A visual guide to the pandemic (BBC press release, 30 March 2020).
- McMullan, L.K., Flint, M., Chakrabarti, A., Guerrero, L., Lo, M.K., Porter, D., Nichol, S.T., Spiropoulou, C.F. and Albariño, C. (2019) Characterisation of infectious Ebola virus from the ongoing outbreak to guide response activities in the Democratic Republic of the Congo: a phylogenetic and in vitro analysis. Lancet Infect Dis., 19(9), 1023–1032.
- Informal consultation on prioritization of candidate therapeutic agents for use in novel coronavirus 2019 infection (WHO R&D Blueprint report, 24 January 2020).
- Gordon, C.J., Tchesnokov, E.P., Feng, J.Y., Porter, D.P. and Gotte, M. (2020) The antiviral compound remdesivir potently inhibits RNA-dependent RNA polymerase from Middle East respiratory syndrome coronavirus. J Biol Chem., in press.
- Holshue, M.L., DeBolt, C., Lindquist, S., Lofy, K.H., Wiesman, J., Bruce, H., Spitters, C., Ericson, K., Wilkerson, S., Tural, A., Diaz, G., Cohn, A., Fox, L., Patel, A., Gerber, S.I., Kim, L., Tong, S., Lu, X., Lindstrom, S., Pallansch, M.A., Weldon, W.C., Biggs, H.M., Uyeki, T.M., Pillai, S.K. and Washington State 2019-nCoV Case Investigation Team. (2020) First case of 2019 novel coronavirus in the United States. N Engl J Med., 382(10), 929–936.
- Gilead Sciences initiates two phase 3 studies of investigational antiviral remdesivir for the treatment of COVID-19 (Gilead Sciences press release, 26 February 2020).
1st March 2020, Halicin
Antibiotics are the cornerstone of modern medicine and since the discovery of penicillin in 1928 by Alexander Fleming (1881–1955), these essential medicines have saved the lives of millions of people worldwide. However, their continued efficacy is uncertain due to the rapid emergence of drug-resistant bacteria which pose a major threat to human health [1,2]. It is estimated that 700,000 people die each year globally due to drug-resistant infections and the number will continue to rise to 10 million by 2050. The overuse and misuse of these medicines drives the evolution of resistance, and the crisis is exacerbated by the lack of development of new antibiotics by the pharmaceutical industry due to challenging regulatory requirements and reduced economic incentives [3].Historically, antibiotics were discovered by screening natural sources such as plant extracts, soil-dwelling microbes, and moulds to identify secondary metabolites that possess antibiotic properties. This approach resulted in the majority of clinically useful chemical classes including the β-lactams. Synthetic antibiotics of the sulfa, pyrimidine and quinolone classes were also discovered later. Unfortunately, the discovery of novel antibiotics has become increasingly difficult [3]. Efforts to discover new broad-spectrum antibiotics using combinatorial chemistry and high throughput screening of large chemical compound collections have failed while mining of soil bacteria identifies the same scaffolds which have been previously discovered. Therefore, novel approaches to antibiotic discovery are urgently required [4].
Now, a group of researchers from Massachusetts Institute of Technology (MIT) have described their use of a machine-learning algorithm to discover a novel broad-spectrum bactericidal antibiotic called halicin (also known as SU-3327, CHEBI:146227). Their work recently published in the journal, Cell, trained a deep neural network on a dataset of 2,560 compounds from a list of FDA-approved drugs and natural products found in plants, animals and microbes to predict molecules capable of inhibiting the growth of the bacterium Escherichia coli [3]. Once the model was trained, they tested the model against a diverse library of more than 6,000 molecules at various stages of investigation for human diseases and identified 99 compounds which were predicted by the model to display antibacterial properties. These were subsequently tested experimentally, and 51 molecules displayed growth inhibition against E. coli, one of which was the kinase inhibitor, halicin, originally developed for the treatment of diabetes and structurally dissimilar to existing antibiotics [3,5].
Tests in cells showed that halicin displayed potent inhibitory activity against a range of antibiotic-resistant strains of bacteria including E. coli, Mycobacterium tuberculosis and C. difficile. The researchers also tested the compound in mice infected with a specific strain of Acinetobacter baumannii, an aggressive pathogen commonly found in hospitals which is resistant to all known antibiotics. Preliminary studies suggest that halicin kills bacteria through the disruption of their ability to maintain an electrochemical gradient across cell membranes. The team subsequently used their model, to screen over 107 million virtual molecules from the ZINC15 database over the course of 4 days and found 23 potential candidates, of which 8 possessed antibacterial activity. This study is a great example of how artificial intelligence can be used to speed up early-stage drug discovery, saving both time and money [3].
The background image is a detail from a Creative Commons licensed image showing a scanning electron micrograph of Escherichia coli.
Reference(s)
- Ventola, C.L. (2015) The antibiotic resistance crisis, part 1: causes and threats. P&T, 40(4), 277–283.
- Zaman, S.B., Hussain, M.A., Nye, R., Mehta, V., Mamun, K.T. and Hossain, N. (2017) A review on antibiotic resistance: alarm bells are ringing. Cureus, 9(6), e1043.
- Stokes, J.M., Yang, K., Swanson, K., Jin, W., Cubillos-Ruiz, A., Donghia, N.M., MacNair, C.R., French, S., Carfrae, L.A., Bloom-Ackerman, Z., Tran, V.M., Chiappino-Pepe, A., Badran, A.H., Andrews, I.W., Chory, E.J., Church, G.M., Brown, E.D., Jaakkola, T.S., Barzilay, R. and Collins, J.J. (2020) A deep learning approach to antibiotic discovery. Cell, 180(4), 688–702.
- Cox, G., Sieron, A., King, A.M., De Pascale, G., Pawlowski, A.C., Koteva, K. and Wright, G.D. (2017) A common platform for antibiotic dereplication and adjuvant discovery. Cell. Chem. Biol., 24(1), 98–109.
- De, S.K., Stebbins, J.L., Chen, L.H., Riel-Mehan, M., Machleidt, T., Dahl, R., Yuan, H., Emdadi, A., Barile, E., Chen, V., Murphy, R. and Pellecchia, M. (2009) Design, synthesis, and structure-activity relationship of substrate competitive, selective, and in vivo active triazole and thiadiazole inhibitors of the c-Jun N-terminal kinase. J. Med. Chem., 52(7), 1943–1952.
1st February 2020, Kaletra
Coronaviruses are a family of viruses which affect the respiratory tract of humans and includes the well-known severe acute respiratory syndrome coronavirus (SARS-CoV) and Middle East respiratory syndrome coronavirus (MERS-CoV), which have claimed the lives of hundreds of people in the past 17 years. They were given their name based on the crown-like projections on their surfaces - "corona" means "crown" in Latin [1].The first coronavirus was isolated from chicken embryos in 1937 and over the last few decades, scientists have found that coronaviruses can infect several animal species, including mice, rats, cats, dogs, birds, bats, snakes, cattle and humans [2,3]. Typically, humans develop pneumonia-like symptoms including fever, sore throat, dry cough, fatigue and breathing difficulties.
In late December 2019, a new strain of the virus designated 2019-nCoV broke out in China which, at the time of writing, has infected more than 7,700 people and killed 170 [4]. A study of the genetic code of the 2019-nCoV strain reveals that the virus may have originated from the host species, bats, which passed the disease to snakes which in turn passed it to humans [5,6]. The source of the outbreak is believed to be Wuhan seafood market, now shut down, which sold many different animals including snakes and poultry [4].
China's National Health Commission Minister, Ma Xiaowei, has reported that a person with the Wuhan coronavirus could be infectious to others even before any symptoms of their own infection appeared [7]. If this is the case, then stopping the spread of the virus will be very difficult. In an attempt to enforce strict containment measures to curb the spread of the new coronavirus, 56 million people across the country have been placed under lockdown (where most private cars are banned, trains and flights suspended), including in Wuhan, the capital of Hubei province where the outbreak originated [4].
To date, there are no antiviral drugs that specifically target human coronaviruses. As the death toll rises and the global race to find a cure continues, China is currently testing a combination of lopinavir and ritonavir (CHEBI:145924, brand name: Kaletra) sold by AbbVie as part of its latest treatment plan for patients infected by the coronavirus [8]. It is thought that the drug, originally developed for the treatment of human immunodeficiency virus (HIV), may block a protease that the coronavirus needs for reproduction, thereby delaying disease progression.
Kaletra has previously been tested in patients with SARS and MERS and recently, a leading Chinese respiratory doctor who contracted the virus after visiting Wuhan took Kaletra as part of his treatment, and it appeared to be effective [8,9]. This new coronavirus outbreak is another reminder of our vulnerability to emerging viral infections and that people should limit the consumption of wild animals to prevent zoonotic infections.
Despite widespread media hype, the new virus appears to be relatively mild. During the winter of 2008-2009, over 13,000 people died from influenza in the UK alone (despite an annual vaccination campaign). The World Health Organization (WHO) estimates that globally, flu kills between 250,000 and 500,000 people every year [10].
The background image is a detail from a Creative Commons licensed image showing an electron micrograph of coronavirus virions.
Reference(s)
- Fan, Y., Zhao, K., Shi, Z.L. and Zhou, P. (2019) Bat coronaviruses in China. Viruses, 11(3), 210.
- Beaudette, F.R. and Hudson, C.B. (1937) Cultivation of the virus of infectious bronchitis. J. Am. Vet. Med. Assoc., 90, 51–58.
- Fehr, A.R. and Perlman, S. (2015) Coronaviruses: an overview of their replication and pathogenesis. Methods Mol. Biol., 1282, 1–23.
- Coronavirus infection: everything you need to know about the outbreak from China (The Telegraph press release, 30 January 2020).
- Ji, W., Wang, W., Zhao, X., Zai, J. and Li, X. (2020) Homologous recombination within the spike glycoprotein of the newly identified coronavirus may boost cross-species transmission from snake to human. J. Med. Virol., in press.
- Luo, G. and Gao, S.J. (2020) Global health concern stirred by emerging viral infections. J. Med. Virol., in press.
- China coronavirus 'spreads before symptoms show' (BBC press release, 26 January 2020).
- China names HIV drugs in treatment plan for new virus (Bloomberg press release, 26 January 2020).
- Chu, C.M., Cheng, V.C., Hung, I.F., Wong, M.M., Chan, K.H., Chan, K.S., Kao, R.Y., Poon, L.L., Wong, C.L., Guan, Y., Peiris, J.S. and Yuen, K.Y. (2004) Role of lopinavir/ritonavir in the treatment of SARS: initial virological and clinical findings. Thorax., 59(3), 252–256.
- Influenza (flu) (Vaccine knowledge project, 22 November 2019).
2nd January 2020, Nitrogen dioxide
Air pollution is a global health risk that is responsible for millions of premature deaths each year. A new study published in the European Heart Journal estimates that, in Europe alone, nearly 790,000 people die each year due to air pollution, and that each life is cut short by an average of more than 2 years [1].With air quality a serious concern, the Copernicus Sentinel-5P satellite was launched in 2017 as part of the European Union's Earth observation programme to map a multitude of air pollutants around the globe. The satellite carries the most advanced spectrometer to date, the Tropomi (TROPOspheric Monitoring Instrument) which can detect the unique fingerprint of atmospheric gases and can image air pollutants more accurately and at a higher spatial resolution than ever before [2].
One of the first images from the satellite showed levels of nitrogen dioxide (NO2, CHEBI:33101) over Europe. High concentrations of this major air pollutant were seen across parts of the Netherlands, the Po Valley in Italy, the Ruhr area in western Germany, and over parts of Spain. The major source of NO2 is the burning of fossil fuels (coal, oil and gas) and motor vehicle exhaust emissions. Europe's dense population and poor air leads to exposure that is among the highest in the world [2,3].
NO2 belongs to a group of highly reactive gases known as nitrogen oxides (NOx) which contribute to the formation of photochemical smog and the brown sky that often covers large cities. NOx reduce air quality and are an important source of acid rain, particulate matter and ground-level ozone (O3) which can damage ecosystems, plant and animal life. It is well known that NOx can negatively affect vegetation, causing leaf damage and reduced growth, and cause vegetation to become more susceptible to disease [4,5].
NO2 levels exceed air quality standards in many European cities. Increased levels of NO2 irritate and inflame the lining of airways and exacerbate asthma, COPD, coughing and breathing problems. Long-term exposure to the gas can affect lung function, increase the likelihood of respiratory problems, and lead to an increase in hospital admissions and mortality [6]. It is suggested that switching to clean renewable energy sources would decrease air pollution-related deaths in Europe by about 55% [1].
The background image is a detail from a Creative Commons licensed picture showing photochemical smog over Mexico City.
Reference(s)
- Lelieveld, J., Klingmüller, K., Pozzer, A., Pöschl, U., Fnais, M., Daiber, A. and Münzel, T. (2019) Cardiovascular disease burden from ambient air pollution in Europe reassessed using novel hazard ratio functions. European Heart Journal, 40(20), 1590–1596.
- Sentinel-5P brings air pollution into focus (European Space Agency press release, 1 December 2017).
- Air pollution deaths are double previous estimates, finds research (The Guardian press release, 12 March 2019).
- Jaffe, L.S. (1967) The biological effects of photochemical air pollutants on man and animals. Am. J. Public Health Nations Health, 57(8), 1269–1275.
- Katz, M. and Shore, V.C. (1955) Air pollution damage to vegetation. J. Air. Pollut. Control Assoc., 5(3), 144–182.
- Kim, K.H., Jahan, S.A. and Kabir, E. (2013) A review on human health perspective of air pollution with respect to allergies and asthma. Environ Int., 59, 41–52.
2019

1st December 2019, Ketamine
Ketamine (CHEBI:6121), commonly used as its hydrochloride salt, brand name: Ketalar, is a potent anaesthetic and analgesic drug which has been on the World Health Organization's List of Essential Medicines since 1985. Its history starts with phencyclidine (also known as angel dust), an anaesthetic drug first synthesised in 1956 by chemists at Parke Davis company. Phencyclidine produced mind-altering effects when tested in animals, including the appearance of drunkenness in rodents, cataleptoid states in pigeons, delirium in dogs, and anaesthesia in monkeys. Although phencyclidine was a safe and reliable anaesthetic in humans, it caused an intense and prolonged-emergence delirium which made the drug undesirable for human use. Therefore, efforts were focussed towards synthesising short-acting analogues of phencyclidine which would have similar anaesthetic potential and a reduced emergence delirium [1,2].Ketamine, originally known by its research code CI-581, was one of many structural analogues synthesised by Parke Davis chemist, Calvin Stevens (1923–2014) in 1962. With one-tenth of the potency of its parent drug phencyclidine, ketamine was subsequently selected for human trials. The first anaesthetic dose was administered to humans in 1964. It was found to be safe and effective as a clinical anaesthetic with minimal side effects and reduced emergence delirium compared to phencyclidine. The drug could rapidly produce profound analgesia with a unique state of altered consciousness and a limited duration of effect which could be prolonged with repeated administration [2]. Ketamine was subsequently approved for human use in 1970 by the Food and Drug Administration (FDA) and was extensively used during the Vietnam War for surgical anaesthesia of wounded soldiers. The mechanism of ketamine's effects is mainly related to its inhibition of the N-methyl-D-aspartate (NMDA) receptors [3]. Ketamine is still widely used as an anaesthetic medicine in veterinary practice. In recent years, however, it has also become a commonly abused ''recreational'' drug, due to its hallucinogenic, tranquilizing and dissociative effects. Sold under its street names of ''K'', ''Special K'' or ''Vitamin K'', it provides feelings of detachment from one's body, which have made it a commonly-used ''date rape drug'' [4].
The background image is a detail from a Creative Commons licensed picture showing ketamine vials ready for administration.
Reference(s)
- Gao, M., Rejaei, D. and Liu, H. (2016) Ketamine use in current clinical practice. Acta. Pharmacol. Sin., 37(7), 865–872.
- Li, L. and Vlisides, P.E. (2016) Ketamine: 50 years of modulating the mind. Front. Hum. Neurosci., 10, 612.
- Domino, E.F. (2010) Taming the ketamine tiger. Anesthesiology, 113(3), 678–684.
- Li, J-H, Vicknasingam, B., Cheung, Y-W., Zhou, W., Nurhidayat, A.W., Jarlais, D.C.D. and Schottenfeld, R. (2011) To use or not to use: an update on licit and illicit ketamine use. Subst. Abuse Rehabil., 2(1), 11–20.
1st November 2019, Cellulose acetate
Cellulose acetate (CHEBI:145232) is a synthetic polymer which was first produced in 1865 by the French chemists, Paul Schützenberger (1829–1897) and Laurent Naudin of the Collège de France, Paris by treatment of cellulose (a naturally occurring polysaccharide found in plants) with acetic anhydride [1].During the outbreak of the First World War, cellulose acetate dope was used for weather proofing and stiffening the fabric of aircraft wings [2]. In 1921, the British Cellulose Co. Ltd. started the first commercial production of acetate filament. They were followed by DuPont in the United States in 1929 with the production of acetate fibre and acetate fabrics were soon in demand for their resistance to staining and creasing, fast-drying and softness [1].
Depending on the way it is processed, cellulose acetate can be used in a variety of applications. As a base material for photographic film, it changed the motion-picture industry by making it possible to store images on a substance which, unlike its cellulose nitrate-based predecessor, doesn't have the tendency to burst into flames. It has also found applications in clothing, membrane filters, coatings, food packaging, and as a frame material for eyeglasses [3].
When concerns due to the association of cigarette smoking with lung cancer emerged in the 1950's, the tobacco industry introduced filter-tipped cigarettes as a means to allegedly make cigarettes safer by absorbing some of the tar which was implicated in the lung cancer epidemic (in fact, the filter tips filter out very little tar; the brown staining of a cigarette filter is due to a change in pH). These filters were (and still are) composed of cellulose acetate which can take a decade or more to decompose [4].
Today, cigarette butts and the cellulose acetate they contain are the leading source of plastic waste worldwide. An estimated 4.5 trillion cigarette butts are discarded annually, representing 22-46% of visible litter. Around 130 chemicals have been reported to be present in used cigarette butts which can leach into the ground and waterways, damaging living organisms that come into contact with them [5].
From 2021, single-use plastics products such as plates, cutlery and straws will be banned by the European Union. These measures were taken due to the growing awareness of the harm done to ecosystems via the disposal of large quantities of plastic into the environment. However, such measures do not extend to the cigarette butt, possibly an indication of the lobbying power of the tobacco industry [4].
The background image is a detail from a Creative Commons licensed picture showing a new and used cigarette filter.
Reference(s)
- Chen, J. (2015) Chapter 4 - Synthetic textile fibers: Regenerated cellulose fibers. Textiles and Fashion. Woodhead Publishing, 79–95.
- Park, J. and Shore, J. (1999) Dye and fibre discoveries of the twentieth century. Part 1: From the magic of electric light to the nightmare of world war. JSDC, 115, 157–167.
- Ciliberto, E., Gemmellaro, P., Iannuso, V., La Delfa. S., Urso, R.G. and Viscuso, E. (2013) Characterization and weathering of motion-picture films with support of cellulose nitrate, cellulose acetate and polyester. Procedia Chemistry, 8, 175–184.
- van Schalkwyk, M.C.I., Novotny, T.E. and McKee, M. (2019) No more butts. BMJ, 367, I5890.
- Benavente, M.J., Caballero, M.J.A., Silvero, G., López-Coca, I. and Escobar, V.G. (2018) Cellulose acetate recovery from cigarette butts. Proceedings, 2, 1447.

1st October 2019, Sulfur hexafluoride
Sulfur hexafluoride (abbreviated as SF6, CHEBI:30496) is a colourless, odourless and non-flammable gas first discovered in 1900 by the French chemists, Henri Moissan (1852–1907) – the discoverer of fluorine – and Paul Lebeau (1868–1959) [1]. In 1937, the General Electrical Company realised that SF6 gas could be used as a gaseous electrical insulator. In the 1960's, the gas was being used in the electrical industry for the prevention of electrical accidents (mainly short circuits and fires) [2].In the past few decades, as the demand for renewable energy has increased, so more connections have been required to the electricity grid, resulting in the increased use of electrical switches and circuit breakers needed to prevent serious accidents. SF6 was widely used to keep this switchgear safe from arcing and short circuits. Today, the gas is still commonly used as an effective insulating material in medium and high-voltage electrical installations, including in large power stations, wind turbines and electrical substations. Its use is also increasing in several other industries, including automobile, health care and semi-conductor manufacturing [3].
The significant downside is that the concentration of SF6 in the Earth's atmosphere is increasing rapidly due to leaks in the electrical industry. Once in the atmosphere, it remains there for millenia (up to 3200 years). SF6 is also an effective absorber of infrared radiation and this combination of properties makes it the World's most potent greenhouse gas, being 23,500 times more warming than carbon dioxide [4].
In 2014, the European Commission tried to prohibit SF6 which belongs to the group of human-produced gases known as fluorinated- or F-gases but faced strong opposition from several lobbying groups. Experts say that for high-voltage applications, there are very few alternatives to SF6 and once viable alternatives do become available, SF6 usage will decline as a result. In 2017, it is estimated that leaks of the gas in the United Kingdom and Europe were the equivalent (in global warming terms) of putting an extra 1.3 million cars on the road. The UK's energy regulator Ofgem is trying to work with energy companies to limit leaks of the gas [3].
The background image is a detail from a Creative Commons licensed picture showing an SF6 circuit breaker installed at a hydroelectric generating station.
Reference(s)
- Moissan, H. and Lebeau, P.C.R. (1900) Sur un nouveau corps gazeux: Le perfluorure de soufre SF6. Acad. Sci. Paris C. R. , 130, 865–871.
- Xiao, S., Zhang, X., Tang, J. and Liu, S. (2018) A review of SF6 substitute gases and research status of CF3I gases. Energy Reports, 4, 486–496.
- Climate change: Electrical industry's 'dirty secret' boosts warming (BBC press release, 13 September 2019).
- Dervos, C.T. and Vassiliou, P. (2000) Sulfur hexafluoride (SF6): global environmental effects and toxic byproduct formation. J. Air Waste Manage. Assoc., 50(1), 137–141.

1st September 2019, Helium hydride ion
When the universe first formed 13.8 billion years ago, only a few types of atoms existed (mainly hydrogen and helium). It was long hypothesised that the first chemical compound in the universe was the helium hydride ion (HeH+, CHEBI:33688). Destruction of HeH+ led to the formation of molecular hydrogen (H2), the most significant and abundant molecule in the universe and which provided a dominant mechanism for gas to cool and form stars, thereby influencing the structure of the early universe [1,2].In 1925, scientists were able to produce HeH+ in the laboratory, using electron impact ionisation [3]. In the 1970's, astronomers searched for HeH+ in giant clouds of dust and gas known as nebulae, especially planetary nebulae which could mimic conditions of the early universe. However, their hunt was unsuccessful [2].
Until very recently, HeH+ was difficult to detect in interstellar space and so its existence has been a dilemma for astronomy for decades. A major problem was that the Earth's atmosphere absorbs the infrared signature given off by these ions.
To tackle this issue, scientists used the largest airborne observatory in the world – an 80/20 joint project of NASA and the German Aerospace Center (DLR) known as the Stratospheric Observatory for Infrared Astronomy (SOFIA) – an observatory aboard a modified Boeing 747SP aircraft (SP = Special Performance). Flying at an altitude of over 40,000 feet (12 km), it is above 99% of the Earth's infrared-blocking atmosphere, so enabling the scientists to detect HeH+ for the first time in the planetary nebula NGC 7027, located 3,000 light-years from Earth [4].
The remarkable discovery marks the culmination of a decades-long search for these ions and helps to confirm our understanding of chemistry in the evolution of the early universe. Details have recently been published in the journal Nature [4].
The background image is a Creative Commons licensed image of the planetary nebula, NGC 7027 which was taken by the Hubble space telescope.
Reference(s)
- First astrophysical detection of a very special molecule (SOFIA press release, 17 April 2019).
- The universe's first type of molecule is found at last (NASA press release, 17 April 2019).
- Hogness, T.R. and Lunn, E.G. (1925) The ionization of hydrogen by electron impact as interpreted by positive ray analysis. Phys. Rev., 26(1), 44–55.
- Güsten, R., Wiesemeyer, H., Neufeld, D., Menten, K.M., Graf, U.U., Jacobs, K., Klein, B., Ricken, O., Risacher, C. and Stutzki, J. (2019) Astrophysical detection of the helium hydride ion HeH+. Nature, 568(7752), 357–359.
1st August 2019, trans-Resveratrol
Fifty years after first landing a man on the moon, NASA is planning to send astronauts to Mars in the near future. Gravity on Mars is 62% lower than it is here on Earth – a human who weighs 100 kg on Earth would weigh just 38 kg on the red planet. In this low-gravity environment, muscles and bones in the human body would weaken. One of the biggest challenges is to ensure that the first adventurers are in good shape and able to carry out their tasks upon arrival on Mars as astronauts en route to Mars won't have the type of exercise machines deployed on the International Space Station [1].A recent study carried out by researchers at Harvard Medical School suggests that trans-resveratrol (CHEBI:45713) [2], a naturally occurring polyphenol commonly found in peanuts, grapes, blueberries, pomegranates and red wine, may hold the key to help prevent muscle deconditioning in low gravity environments [1,3].
After 3 weeks in space, the soleus calf muscle shrinks by a third, followed by the loss of slow-twitch muscle fibres which are necessary for endurance. The researchers found that trans-resveratrol can preserve muscle and bone mass in rats without impacting food intake or total body weight. Although the reasons for the effect of trans-resveratrol on the muscles is not well understood, it is suggested that the compound maintains glucose uptake in the muscles, allowing them to perform better [1,4].
trans-Resveratrol is widely used as a dietary supplement and was first isolated in 1940 from the roots of the white hellebore plant, Veratrum grandiflorum [5]. It has been extensively investigated for its anti-oxidative, anti-inflammatory, anti-hypertensive and anti-diabetic properties [3,6], though many of the health claims are not without controversy.
The background image is a detail from a Creative Commons licensed picture showing the surface of Mars.
Reference(s)
- Mortreux, M., Riveros, D., Bouxsein, M.L. and Rutkove, S.B. (2019) A moderate daily dose of resveratrol mitigates muscle deconditioning in a Martian gravity analog. Front. Physiol., 10(899), 1–6.
- Entity of the Month 1 November 2015, trans-resveratrol.
- Fremont, L. (2000) Biological effects of resveratrol. Life Sciences, 66(8), 663–673.
- An antioxidant in red wine might power astronauts on Mars, study says (CNN press release, 19 July 2019).
- Takaoka, M. (1940) Phenolic substances of white hellebore (Veratrum grandiflorum Loes. fil.). J. Faculty Sci., Hokkaido Imp. Univ., 3(Ser. III), 1–16.
- de Sá Coutinho, D., Pacheco, M.T., Frozza, R.L. and Bernardi, A. (2018) Anti-inflammatory effects of resveratrol: Mechanistic insights. Int. J. Mol. Sci., 19(6), E1812.

1st July 2019, Cocaine
Cocaine (CHEBI:27958) is a natural alkaloid that was first isolated in 1859 by the German chemist, Albert Niemann (1834–1861) from the leaves of the Erythroxylum coca shrub found in South America [1]. For centuries, natives of this region consumed the drug by chewing coca leaves to boost energy, combat fatigue resulting from working at high altitudes and stave off hunger [2].In 1884, Carl Koller (1857–1944), an Austrian ophthalmologist, successfully used cocaine as a local anaesthetic in a cataract operation [2]. By the early 1900s, cocaine was commonly being used as an ingredient in many non-prescription medicines and tonics and was also an ingredient in the original Coca-Cola soft drinks formula [3,4].
However, the United States (US) banned the drug in 1914 after its harmful and addictive effects became apparent, and its medical use dramatically decreased thereafter [3]. Today, cocaine is still used by some physicians as a topical anaesthetic during nasal surgery due to its vasoconstrictive and numbing properties [5]. Cocaine addiction is a major global public health problem. Just last month (June 2019), US customs seized 16 tons of cocaine with an estimated street value of more than $1bn from a ship docked in Philadelphia – considered one of the largest drug busts in US history [6].
A potent central nervous system (CNS) stimulant, cocaine delivers a powerful high. Years of research have suggested that it exerts its psychoactive effects by binding to dopamine transporters, so inhibiting the removal of the neurotransmitter dopamine from the synapse. The consequent increase in the levels of dopamine in the synapse results in an amplified signal to the receiving neurons which is responsible for the high feeling or euphoria. Cocaine also increases the levels of other neurotransmitters (norepinephrine and serotonin) in the brain. The overall effect results in increased heart rate and blood pressure, vasoconstriction and increases the risk of having a stroke or heart attack [7,8]. In 2017, there were an estimated 14,000 deaths in the US from cocaine use – an increase of 34% in a single year [9].
The background image is a detail from a Creative Commons licensed picture showing the leaves of the Erythroxylum coca shrub.
Reference(s)
- Niemann, A. (1860) Ueber eine neue organische Base in den Cocablättern. Archiv der Pharmacie, 153(2), 129–155.
- McLaughlin, G.T. (1973) Cocaine: The History and regulation of a dangerous drug. Cornell Law Review, 58(3), 537–573.
- Brain, P.F. and Coward, G.A. (1989) A review of the history, actions, and legitimate uses of cocaine. J. Subst. Abuse, 1(4), 431–451.
- Das, G. (1993) Cocaine abuse in North America: A milestone in history. J. Clin. Pharmacol., 33(4), 296–310.
- Harper, S.J. and Jones, N.S. (2006) Cocaine: what role does it have in current ENT practice? A review of the current literature. J. Laryngol. Otol., 120(10), 808–811.
- More than $1bn worth of cocaine seized from ship at Philadelphia port (The Guardian press release, 18 June 2019).
- Nestler, E.J. (2005) The neurobiology of cocaine addiction. Sci. Pract. Perspect., 3(1), 4–10.
- Treadwell, S.D. and Robinson, T.G. (2007) Cocaine use and stroke. Postgrad. Med. J., 83(980), 389–394.
- Kariisa, M., Scholl, L., Wilson, N., Seth, P. and Hoots, B. (2019) Drug overdose deaths involving cocaine and psychostimulants with abuse potential – United States, 2003-2017. Morb. Mortal. Wkly. Rep., 68(17), 388–395.

1st June 2019, Neopentyl glycol
Worldwide power consumption for refrigeration and air conditioning (A/C) is growing rapidly. As incomes and living standards improve in countries such as China and India, so the demand for A/C is set to rise. It is estimated that worldwide power consumption for A/C alone will increase 33-fold by the year 2100 and energy used for cooling could exceed that used for heating within the next 30 years [1,2].Hydrofluorocarbons (HFCs) are frequently used as refrigerants in refrigerators and A/C units and were commercialised in the 1990s as substitutes for the ozone-depleting chlorofluorocarbons and hydrochlorofluorocarbons [3]. However, these are toxic, flammable and typically 4,000 times more potent as greenhouse gases than carbon dioxide (CO2). They can leak into the atmosphere when refrigerators and A/C units are manufactured, installed and discarded and have a high global warming potential [1]. According to the US Department of Energy, three out of every four homes in the US have air conditioners which emit nearly 117 million metric tons of CO2 into the atmosphere each year. Therefore, there is an urgent need to find safer alternatives to HFCs [4].
Recently, scientists from the University of Cambridge together with collaborators in Spain have identified neopentyl glycol (CHEBI:143768), as an eco-friendly and inexpensive solid which could replace HFCs in refrigerators and air conditioners [5]. This material is widely used as an additive in the synthesis of cosmetics, paints and lubricants. Their research published in the journal Nature Communications shows that plastic crystals of neopentyl glycol can absorb heat from the external environment and deliver a cooling effect comparable to that observed with HFC's when compressed. This is achieved by a change in the material's microscopic structure and can pave the way for the development of more safer, greener and efficient cooling systems [6].
The background image is a detail from a Creative Commons licensed image showing a wall mounted air conditioning condenser.
Reference(s)
- World set to use more energy for cooling than heating (The Guardian press release, 26 October 2015).
- How America became addicted to air conditioning (The Guardian press release, 26 October 2015).
- McLinden, M.O., Brown, J.S., Brignoli, R., Kazakov, A.F. and Domanski, P.A. (2017) Limited options for low-global-warming-potential refrigerants. Nat Commun., 8, 14476.
- Air conditioning (The U.S. Department of Energy).
- Green material for refrigeration identified (The University of Cambridge press release, 18 April 2019).
- Lloveras, P., Aznar, A., Barrio, M., Negrier, P., Popescu, C., Planes, A., Mañosa, L., Stern-Taulats, E., Avramenko, A., Mathur, N.D., Moya, X. and Tamarit, J.L. (2019) Colossal barocaloric effects near room temperature in plastic crystals of neopentylglycol. Nat Commun., 10(1), 1803.
1st May 2019, Glyphosate
Glyphosate (CHEBI:27744, trade name: Roundup), is an organophosphorus compound discovered by a team of scientists led by Dr John Franz and brought to market for agricultural use in 1974 by Monsanto. It is the most widely used systemic broad-spectrum herbicide and crop desiccant in the US; in 2014, total worldwide consumption of glyphosate stood at 826 million kg [1]. After its patent expired in 2000, the chemical began to be sold by several manufacturers and is a constituent in more than 750 products being sold in the US [2]. Despite this competition, Monsanto is still a dominant player in the market and focusses its strategy on genetically modified (GM) glyphosate resistant crops.Glyphosate is used by farmers to kill weeds, especially annual broadleaf weeds and grasses that compete with crops like corn, soybeans, and cotton. It works by interfering with the shikimate pathway which in plants produces the necessary aromatic amino acids phenylalanine, tyrosine and tryptophan. The herbicide inhibits the plant enzyme 5-enolpyruvylshikimate-3-phosphate synthase resulting in plant death from lack of amino acids the plant requires to survive. Plant growth stops within hours of application, whilst the leaves can take several days to turn yellow [3].
Although it is used globally as a safe and effective means of weed control, glyphosate continues to be in the headlines. A US jury found that glyphosate was a substantial factor in causing non-Hodgkinâs lymphoma in a California resident [4]. The pharmaceutical company Bayer which acquired Roundup as part of its $66 billion takeover of US rival Monsanto strongly rejected these claims. Such claims however are not new; there are 11,200 Roundup lawsuits still pending in the US. In August, a state court jury awarded $289 million to another California resident after finding that long term exposure to Roundup caused his cancer, sending Bayer shares plunging at the time [4].
In March 2015, the cancer-research arm of the World Health Organisation (WHO) classified glyphoshate as 'probably carcinogenic to humans'. However, the US environmental protection agency (EPA) insist that it is safe when used carefully. Glyphosate use has been banned in public parks and gardens in some countries and regions [5,6].
The background image is a detail from a Creative Commons licensed picture showing a typical spectral signature of cereal plants treated with the herbicide glyphosate.
Reference(s)
- Benbrook, C.M. (2016) Trends in glyphosate herbicide use in the United States and globally. Environ. Sci. Eur., 28(3), 1–15.
- Chang, E.T. and Delzell, E. (2016) Systematic review and meta-analysis of glyphosate exposure and risk of lymphohematopoietic cancers. J. Environ. Sci. Health B., 51(6), 402–434.
- Schönbrunn, E., Eschenburg, S., Shuttleworth, W.A., Schloss, J.V., Amrhein, N., Evans, J.N. and Kabsch, W. (2001) Interaction of the herbicide glyphosate with its target enzyme 5-enolpyruvylshikimate 3-phosphate synthase in atomic detail. Proc. Natl. Acad. Sci. USA., 98(4), 1376–1380.
- Weedkiller glyphosate a 'substantial' cancer factor (BBC press release, 20 March 2019).
- W.H.O. report links ingredient in Roundup to cancer (The New York Times press release, 20 March 2015).
- Weedkiller cancer ruling: What do we know about glyphosate? (BBC press release, 11 August 2018).

1st April 2019, Brexanolone
Postpartum depression (PPD), also known as postnatal depression, is a major depressive disorder associated with childbirth. It affects 1 in every 8 women within a month of giving birth and is the leading cause of maternal suicide. Symptoms may include persistent feeling of sadness and anxiety, low mood, loss of interest in her new born, difficulty sleeping, exhaustion, loss of appetite, lack of concentration, irritability, poor self-esteem and suicidal thoughts. In the US, it is estimated that 500,000-750,000 women will suffer with PPD each year [1].Untreated PPD can have a negative effect on family life and can pose serious risks to child development, affecting the behavioural, emotional, cognitive, and physical development of the child and increasing their risk of anxiety or depressive symptoms later in life [2]. To date, new mothers experiencing PPD have been prescribed antidepressants such as selective serotonin reuptake inhibitors which are used for treating depression in the general population. However, these drugs do not achieve adequate response or full remission of symptoms and can sometimes take weeks to months to be effective [3].
Neurosteroids play an important role in the pathophysiology of PPD. Allopregnanolone, a metabolite of the sex hormone progesterone, is a potent positive allosteric modulator of synaptic and extra-synaptic GABAA receptors and is postulated to have a role in triggering PPD. Throughout pregnancy, the plasma concentrations of allopregnanolone rise in concert with progesterone levels. After childbirth, these concentrations decrease rapidly. Fluctuations in allopregnanolone levels can have a profound effect on brain function. In mice, depressive-like behaviours were reversed upon administration of allopregnanolone. Unfortunately, endogenous allopregnanolone has poor oral bioavailability and low aqueous solubility and is rapidly metabolized [2].
Brexanolone (CHEBI:50169, brand name: Zulresso), a proprietary formulation of allopregnanolone was developed by Sage Therapeutics to overcome these issues and was granted Breakthrough Therapy Designation status in 2016. In clinical studies, many women with moderate to severe PPD saw a marked improvement of their symptoms within just 24 hours of receiving the drug and this response was sustained for up to 30 days post-treatment. The drug is delivered under medical supervision intravenously through a 60-hour infusion [3].
In March 2019 brexanolone was approved by the U.S. Food and Drug Administration (FDA), becoming the first and only drug to be approved specifically for the treatment of PPD. Financial analysts are predicting annual sales of $100 million in its first full year on the market [4].
The background image is a detail from a Creative Commons licensed picture showing a depressed mother with a baby.
Reference(s)
- Kose, S. and Cetin, M. (2017) Brexanolone: an allosteric modulator of GABA-A receptors in the rapid treatment of postpartum depression. Psychiatry and Clinical Psychopharmacology, 27(4), 326–328.
- Kanes, S., Colquhoun, H., Gunduz-Bruce, H., Raines, S., Arnold, R., Schacterle, A., Doherty, J., Epperson, C.N., Deligiannidis, K.M., Riesenberg, R., Hoffmann, E., Rubinow, D., Jonas, J., Paul, S. and Meltzer-Brody, S. (2017) Brexanolone (SAGE-547 injection) in post-partum depression: a randomised controlled trial. Lancet, 390(10093), 480–489.
- Meltzer-Brody, S., Colquhoun, H., Riesenberg, R., Epperson, C.N., Deligiannidis, K.M., Rubinow, D.R., Li, H., Sankoh, A.J., Clemson, C., Schacterle, A., Jonas, J. and Kanes, S. (2018) Brexanolone injection in post-partum depression: two multicentre, double-blind, randomised, placebo-controlled, phase 3 trials. Lancet, 392(10152), 1058–1070.
- This postpartum-depression drug has unusual potential (Bloomberg press release, 20 March 2019).

1st March 2019, Ibuprofen
This month's entity of the month is in recognition of the British pharmacist, Dr Stewart Adams (1923–2019) for his contribution to the discovery of one of the most widely used painkillers, ibuprofen (CHEBI:5855). After leaving school at the age of 16, Stewart Adams started an apprenticeship in retail pharmacy at Boots and later gained a degree in pharmacy from University College Nottingham (now known as the University of Nottingham). In 1952, after completing his PhD in pharmacology at the University of Leeds, he returned to the research department at Boots Pure Drug Company Ltd and was assigned to a project to search for new treatments for Rheumatoid Arthritis (RA), an autoimmune disorder which causes severe swelling, pain and restricted movement in joints. At the time, corticosteroids and high doses of aspirin were commonly used to treat the condition but unpleasant side effects such as stomach irritation, indigestion and bleeding meant that they were far from ideal [1].Aspirin, developed in 1897 was the main painkiller on the market at the time and was the first Non-Steroidal Anti-Inflammatory Drug (NSAID). Dr Adams started to investigate how aspirin worked and set out to find a drug that would be better tolerated than aspirin. He was searching for a novel NSAID that would be 'chemically' non-steroidal, possess improved efficacy over aspirin in reducing pain and inflammation in the joints and in alleviating fevers and flu-like symptoms, without significant side effects [1].
Over the next 10 years, Dr Adams and his team synthesised and tested over 600 chemical compounds. Four of these entered clinical trials but three failed either due to lack of efficacy or unacceptable side effects. Ibufenac, the fourth compound, did make it to the market in 1966 but was later withdrawn due to liver toxicity after prolonged dosing. Eventually, the team discovered a new lead compound which had reasonable potency and a promising safety profile. This was 2-(4-isobutylphenyl) propionic acid, later to become widely known as ibuprofen. Clinical testing showed that the drug had a good safety margin and was effective in treating patients with rheumatoid arthritis [1,2]. Ibuprofen is administered as a racemic mixture, where the R-enantiomer undergoes extensive interconversion to the active S-enantiomer in vivo and exerts its anti-inflammatory action by inhibiting the cyclooxygenase (COX) enzymes 1 and 2 which are involved in prostaglandin synthesis. Prostaglandins are important mediators of inflammation, and by lowering the levels of prostaglandins, ibuprofen can reduce inflammation, pain and fevers [3].
A patent for the drug was granted to Boots in 1962 and ibuprofen was licensed as a prescription drug in 1969 for the treatment of rheumatic diseases. After extensive trials on a range of non-rheumatic painful conditions, the drug was eventually approved as an over-the-counter medicine in 1983. Today, ibuprofen is included in the WHO's Model List of Essential Medicines and used by millions of people around the world for the treatment of fever, mild headaches, rheumatoid arthritis, muscle aches and menstrual pain [1,2].
The background image is a detail from a Creative Commons licensed photograph of a bottle of ibuprofen tablets with cap removed and tablets in front.
Reference(s)
- Halford, G.M., Lordkipanidzé, M. and Watson, S.P. (2012) 50th anniversary of the discovery of ibuprofen: An interview with Dr Stewart Adams. Platelets, 23(6), 415–422.
- Rainsford, K.D. (2011) Fifty years since the discovery of ibuprofen. Inflammopharmacol., 19(6), 293–297.
- Evans, A.M. (2001) Comparative pharmacology of S(+)-ibuprofen and (RS)-ibuprofen. Clin. Rheumatol., 20(1), S9–14.

1st February 2019, Main Group Element Atom
Throughout the 19th Century, chemists tried to organise the known elements so as to reflect similarities in their properties. In 1864, the British chemist John Newlands (1837–1898) arranged the elements in order of increasing atomic weight. He noticed that elements with similar physical and chemical properties occurred after each interval of seven elements (the inert gases were still unknown). He likened this periodicity to the musical scale, but his "Law of Octaves" was initially ridiculed [1].In 1869, the Russian chemist, Dimitri Mendeleev (1834–1907) presented his findings to the Russian Chemical Society. He had also arranged the elements in order of increasing atomic weight, but did not 'force' the periodicity – if an element appeared to be in the wrong place when positioned by atomic weight, he moved it to where it fitted the pattern that he had discovered. Furthermore, he left gaps in his table where it appeared that there should be an element which had not yet been discovered. He even predicted the properties of five of these elements and their compounds. His Periodic Table has been hailed as chemistry's most important breakthrough [2].
The table that hangs in chemistry classrooms around the world today is a direct descendant of the table that Mendeleev drafted in 1869 and his basic organizing principles are still followed. The modern periodic table has vertical columns called groups and horizontal rows called periods. Elements in the same group have similar chemical properties since they have the same number of outer electrons and valency. Elements in a specific period have the same number of atomic orbitals. The number of electrons increases as one moves across and down the table.
During the time of Mendeleev, only 63 elements were known. Changes in the periodic table became necessary as new elements were isolated. In 1894, William Ramsay (1852–1916) discovered the inert gas argon. This behaved like no other known gas since it was totally unreactive. Since Ramsay understood the principles of Mendeleev's table and knew that elements were sorted into groups, argon was put into a new group, then known as group 0. Over the next few years, several new members of this group were discovered and the group became known as the inert or noble gases [3].
Although Mendeleev never received a Nobel Prize for his work, the element Mendelevium (atomic number 101) was named after him, in recognition of his work in the field [2]. This month, both Mendeleev and his table made headlines when the world's oldest surviving periodic table – dating back as far as 1879 – was discovered during a laboratory clear-out at the University of St Andrews [4].
Since its inception, the periodic table and the elements contained within it have shaped our lives. The modern technologies we use on a daily basis employ elements in increasing quantities. The latest mobile phones contain as many as 70 of the 118 elements in the current periodic table. For example, lithium is used in the phone's battery, silicon and aluminium is used in the screen, gold is used in the circuit board, tin in used in solder and carbon is used in the case [5].
The United Nations have designated 2019 as the "International Year of the Periodic Table" marking the 150th anniversary of Mendeleev's achievement [6].
The background image is a detail from a Creative Commons licensed image of a modern periodic table.
Reference(s)
- Giunta, C.J. (1999) J. A. R. Newlands' classification of the elements: periodicity, but no system. Bull. Hist. Chem., 24, 24–31.
- Development of the periodic table (Royal Society of Chemistry).
- Rayleigh, L. and William, R (1895). Argon: A new constituent of the atmosphere. Philosophical Transactions of the Royal Society A., 186, 187–241.
- Periodic table found at St Andrews University 'is world's oldest' (BBC press release, 17 January 2019).
- Supanchaiyamat, N. and Hunt, A.J. (2019) Conservation of critical elements of the periodic table. ChemSusChem., 12, 1–8.
- The United Nations proclaims the international year of the periodic table of chemical elements (IUPAC press release, 28 December 2017).
2nd January 2019, Azidothymidine
Azidothymidine (CHEBI:10110, also known as AZT or zidovudine) was developed in 1964 as a potential anti-cancer agent by American scientist Dr Jerome Horwitz (1919 – 2012) at the Michigan Cancer Foundation (now the Barbara Ann Karmanos Cancer Institute) in Detroit [1]. AZT is a member of a group of compounds called dideoxythymidines which were designed to resemble nucleosides, the building blocks of DNA. In theory, these synthetic nucleosides would substitute themselves for natural nucleosides, and act as chain-terminators of DNA synthesis, so preventing the cancer cells from duplicating. Unfortunately, AZT was ineffective against cancer when tested in mice and the drug was subsequently shelved and forgotten for almost two decades [2].In 1981, the growing AIDS (acquired immunodeficiency syndrome) pandemic prompted a widespread search for new treatments. A few years later, scientists discovered the virus which causes AIDS, known as the human immunodeficiency virus (HIV) [3]. Drug companies and research laboratories subsequently started testing thousands of compounds in the race to find a treatment for AIDS. Burroughs Wellcome (now known as GlaxoSmithKline) sent a resynthesised version of AZT, then known as compound S, to the National Cancer Institute along with several other compounds to see if it could inhibit HIV replication. In the test tube at least, the drug was able to prevent HIV replication by inhibiting the reverse transcriptase enzyme which is essential for the reproduction of viral DNA [4].
What Horwitz did not know was that the drug which he discovered and abandoned 20 years previously was destined for success. AZT subsequently entered clinical trials and was approved in record time (within 2 years). The trial was stopped early since the mortality rate of patients taking AZT was significantly lower compared with that of those taking the placebo and the need for a treatment outweighed the need for rigorous testing [4]. In 1987, AZT became the first antiretroviral drug to be approved by the Food and Drug Administration (FDA) for the treatment of HIV infection and AIDS [5].
The background image is a detail from a Creative Commons licensed image depicting a red ribbon. It is a universal symbol of awareness and support for people living with HIV and AIDS.
Reference(s)
- Horwitz, J.P., Chua, J. and Noel, M. (1964) The monomesylates of 1-(2'-deoxy-β-D-lyxofuranosyl)thymine. J. Org. Chem., 29(7), 2076–2078.
- Jerome Horwitz, AZT Creator, Dies at 93 (The New York Times press release, 20 September 2012).
- Sharp, P.M. and Hahn, B.H. (2011) Origins of HIV and the AIDS pandemic. Cold Spring Harb. Perspect. Med., 1(1), a006841.
- Broder, S. (2009) The development of antiretroviral therapy and its impact on the HIV-1/AIDS pandemic. Antiviral Res., 85(1), 1–38.
- U.S. approves drug to prolong lives of aids patients (The New York Times press release, 21 March 1987).
2018

1st December 2018, Platinum-iridium alloy
One of the rarer elements in the Earth's crust (~0.01 ppm), platinum is a silvery-white transition metal with atomic number 78 and is located in group 10 of the periodic table (under palladium and sandwiched between iridium and gold). One of the least reactive of the metal elements, it is often found in its elemental form. Its density (21.4 g/cm3) is greater than that of gold (19.3 g/cm3). It was prized by the Mayans for jewellery, but early European explorers regarded it with disdain – it was often present as an impurity when they mined gold. By the 18th Century, however, scientists were taking an interest in the metal. In Jamaica, Charles Wood (1702–1774), an English ironmaster, experimented on a sample that had been smuggled from Cartagena in what is now Columbia. He reported his findings to a relative, a British doctor and scientist named William Brownrigg (1711–1800), around 1741. Brownrigg wrote up Wood's experiments and performed some of his own. He then reported the results to the Royal Society in 1750, pointing out that the new metal had an extremely high melting point (pure platinum melts at 1768 ℃), and was remarkably stable to acid – it is insoluble in hydrochloric acid and nitric acid, although it does dissolve in hot aqua regia (a mixture of the two) [1].Soon, synthetic alloys of platinum were being prepared and investigated. In 1838, a researcher named Gaudin prepared a 90:10 platinum-iridium alloy (Pt-Ir alloy, Ir-Pt alloy,CHEBI:142649) and commented on its lustre, malleability and remarkable resistance to corrosion. When H. Sainte-Claire Deville and H. Debray carried out trials of various Ir-Pt alloys at the French Mint to determine their suitability for coinage, they found them to be even stronger and more resistant to corrosion than pure platinum. The alloys found wider uses than coinage; thus a 15% Ir-Pt alloy was subsequently used in the construction of chemical plant. The result was that in contrast to the original aims of Sainte-Claire Deville and Debray, the alloys commanded higher prices than pure platinum [2].
In the latter part of the 19th Century, physicists were looking for substances that could retain their mass, form, and linear dimensions indefinitely, and so be used to represent their fundamental units of measurement. The properties of Pt-Ir alloy seemed to tick all the boxes, and it was used for the production of the International Prototype Metre (IPM) and the International Prototype Kilogram (IPK).
For the IPM, Johnson Matthey prepared 250 kg of 10% Ir-Pt, which was used to produce 30 bars with a distorted-X-shaped cross-section (named for Henri Tresca (1814–1885) who devised it so as to minimise torsional strain during length comparisons). One of the bars was identical in length to the existing mètre des Archives and became the official IPM at the first General Conference of Weights and Measures in September 1889. The other bars were distributed among the signatory nations of the Metre Convention for use as national standards. Thus the U.S. received bar No. 27, which measured 0.999 9984 m ± 0.2 μm (1.6 μm short of the international prototype) .
The IPM remained the official standard for defining the metre until 1960, when it was replaced by a definition based on a certain number of wavelengths of a particular emission line of krypton-86. (The current definition, based on the distance travelled by light in a vacuum in a certain time period, was adopted in 1983).
For the IPK, Johnson Matthey prepared three standard kilogram cylinders in 1878 from 10% Ir-Pt. Each had a diameter and height both equal to 39 mm (1.5 inches). Forty more replicas were ordered in 1882. The new International Prototype Kilogram was also approved at the 1889 Weights and Measures conference. The standard unit of mass for scientists ever since has been defined as being equal to the mass of the IPK, which is kept in a vault in the International Bureau of Weights & Measures, known by its French language initials BIPM, in Saint-Cloud, on the outskirts of Paris [3].
Six sister copies and ten working copies of the IPK (eight for 'routine' use and two for special use) are also kept at St-Cloud. There are currently 69 National Prototypes (the UK has numbers 18, 81, and 82). However, periodic verifications of the IPK and the official copies that were undertaken in 1948 and 1989 have concluded that their masses are slowly but surely diverging from each other, and that the IPK itself has lost around 50 μg compared with the national prototypes [4]. The reason is not known.
A new definition of the kilogram was clearly needed. Just as the definition of the metre was changed from the distance apart of two reference marks on a single Ir-Pt bar to an invariant physical constant, so a new definition for the kilogram was sought that was based on an invariable constant, enabling the standard to be independently reproduced by labs around the world.
Last month, after decades of work by numerous national measurement institutions, at the 26th meeting of the General Conference of Weights and Measures in Versailles, a new definition of the kilogram was approved. It defines Planck's constant to be exactly 6.62607015 x 10–34 kg m2 s–1 . Changes to the definitions of three additional basic SI units for electric current (ampere), temperature (kelvin) and amount of substance (mole) were also approved. It will mean that for the first time, all measurement units will be defined by natural phenomena rather than by physical artifacts.
The new definitions will take effect on World Metrology Day, 20th May 2019.
The background image is a Creative Commons licensed photograph of of the national prototype kilogram standard replica no. K20 kept by the US government National Institute of Standards and Technology (NIST), Bethesda, Maryland, shown as it is normally stored, inside two glass bell jars.
Reference(s)
- Watson, W. and Brownrigg, W (1750) Several papers concerning a new semi-metal, called platina; communicated to the Royal Society by Mr. Wm. Watson F. R. S. Philos. Trans. R. Soc. London, 46(496), 584–596.
- Darling, A.S. (1960) Iridium platinum alloys. A critical review of their constitution and properties Platinum Met. Rev., 4(1), 18–26.
- Smith, F.J. (1973) Standard kilogram weights. A story of precision fabrication Platinum Met. Rev., 17(2), 66–68.
- Girard, G. (1994) The third periodic verification of National Prototypes of the kilogram (1988–1992) Metrologia, 31(4), 317–336.

1st November 2018, Fentanyl
The long-standing addiction of sectors within the US population to opioid-based painkillers has become even more dangerous over the last few years. Fentanyl (CHEBI:119915), a synthetic opioid first synthesised by Paul Janssen (1926–2003) in 1959 is one of the most powerful painkillers on the market, being around 50–100 times more potent than morphine [1]. Commonly used as its citrate salt (brand name: Abstral), it is prescribed by doctors for the management of pain in cancer patients. The drug is often administered in the form of a tablet, patch, injection, nasal spray, or sometimes in the form of a lollipop. Once administered, the drug has a rapid onset of action, moving through the bloodstream and interacting with opioid receptors, predominantly the opioid μ-receptors in the brain and spinal cord, thereby providing several days of pain relief [2]. However, the drug is being made illicitly and is flooding into communities across the US, delivering a powerful high, often resulting in overdose and death. The straightforward synthesis, low cost and high potency of the drug have been major contributors to its influx into the street-drug market. Fentanyl is often mixed with other drugs such as heroin or with alcohol which amplifies its potency and dangers [3].In 2016, there were 42,249 opioid-related deaths in the US, more than half of which were attributed to fentanyl and its chemically similar analogues [4]. One such analogue is carfentanil, a drug which is used to tranquilize elephants and is 10,000 times more potent than morphine [5].
As the number of Americans dying from drug overdoses continues to rise, Nebraska recently became the first state in the US to use fentanyl to replace thiopental sodium [6] as part of the drug cocktail administered in the execution of a death row inmate. This comes at a time when drug companies continue to prevent their products from being used for capital punishment [7].
The background image is a detail from a Creative Commons licensed photograph of a statue by Wilfried Pas of Dr. Paul Janssen, the founder of Janssen Pharmaceutica who first synthesised fentanyl. It is located near the centre of Beerse, Belgium, where the company has its headquarters.
Reference(s)
- Vardanyan, R.S. and Hruby, V.J. (2014) Fentanyl-related compounds and derivatives: current status and future prospects for pharmaceutical applications. Future Med. Chem., 6(4), 385–412.
- Stanley, T.H. (2014) The fentanyl story. J. Pain, 15(12), 1215–1226.
- Ellis, C.R., Kruhlak, N.L., Kim, M.T., Hawkins, E.G. and Stavitskaya, L. (2018) Predicting opioid receptor binding affinity of pharmacologically unclassified designer substances using molecular docking. PLoS ONE, 13(5), e0197734.
- Jones, C.M., Einstein, E.B. and Compton, W.M. (2018) Changes in synthetic opioid involvement in drug overdose deaths in the United States, 2010-2016. JAMA., 319(17), 1819–1821.
- Shanks, K.G. and Behonick, G.S. (2017) Detection of carfentanil by LC-MS-MS and reports of associated fatalities in the USA. J. Anal. Toxicol., 41(6), 466–472.
- Entity of the Month 9 March 2011, Thiopental sodium.
- Nebraska becomes first state to use fentanyl in execution (NBC News press release, 14 August 2018).
1st October 2018, Osimertinib
Lung cancer is the most common cause of cancer-related death worldwide, accounting for 1.6 million deaths each year. Approximately 85% of patients belong to the histological subtype known as non-small-cell lung cancer (NSCLC) [1]. Around 10-15% of NSCLC patients in the US and Europe have a mutation in the epidermal growth factor receptor (EGFR) [2]. Initially, these patients respond well to treatment with currently-available EGFR tyrosine kinase inhibitors (TKIs) such as gefitinib and erlotinib, which block the cell-signalling pathways that drive the growth of tumour cells [3].However, after 9-11 months, their tumours develop resistance to EGFR TKI treatment leading to disease progression. In 60% of cases, this is caused by the T790M resistance mutation in EGFR (so called because a gatekeeper threonine residue (T) at position 790 in the adenosine triphosphate (ATP) binding site of EGFR is mutated to a methionine residue (M)). Two possible mechanisms of resistance have been proposed. One is that the mutation to the larger methionine residue prevents TKIs accessing the drug binding site. The second is that the mutation increases the affinity of ATP for the binding site, resulting in reduced binding of ATP-competitive TKIs [3,4].
AstraZeneca has been developing osimertinib (CHEBI:90943, commonly used as its mesylate salt, brand name: Tagrisso), a third-generation, irreversible small-molecule EGFR inhibitor designed to inhibit both EGFR-sensitising and EGFR T790M-resistance mutations [5].
The results of a phase III trial recently published in the New England Journal of Medicine found that median progression-free survival of patients taking osimertinib was nearly doubled at 18.9 months compared with 10.2 months for patients taking the normal treatment (erlotinib or gefitinib) [5].
The U.S. Food and Drug Administration (FDA) has now approved osimertinib (Tagrisso) as a first-line treatment for patients with EGFR mutation-positive NSCLC [6]. Financial analysts are predicting annual peak sales of at least $3 billion for Tagrisso in the next few years [7].
The background image is a detail from a Creative Commons licensed illustration depicting the life cycle of a cancer cell.
Reference(s)
- Herbst, R.S., Morgensztern, D. and Boshoff, C. (2018) The biology and management of non-small cell lung cancer. Nature, 553(7689), 446–454.
- Chougule, A., Prabhash, K., Noronha, V., Joshi, A., Thavamani, A., Chandrani, P., Upadhyay, P., Utture, S., Desai, S., Jambhekar, N. and Dutt, A. (2013) Frequency of EGFR mutations in 907 lung adenocarcioma patients of Indian ethnicity. PLoS One, 8(10), e76164.
- Remon, J. and Planchard, D. (2015) AZD9291 in EGFR-mutant advanced non-small-cell lung cancer patients. Future Oncol., 11(22), 3069–3081.
- Zou, B., Lee, V.H.F., Chen, L., Ma, L., Wang, D.D. and Yan, H. (2017) Deciphering mechanisms of acquired T790M mutation after EGFR inhibitors for NSCLC by computational simulations. Sci. Rep., 7(1), 6595.
- Soria, J.C., Ohe, Y., Vansteenkiste, J., Reungwetwattana, T., Chewaskulyong, B., Lee, K.H., Dechaphunkul, A., Imamura, F., Nogami, N., Kurata, T., Okamoto, I., Zhou, C., Cho, B.C., Cheng, Y., Cho, E.K., Voon, P.J., Planchard, D., Su, W.C., Gray, J.E., Lee, S.M., Hodge, R., Marotti, M., Rukazenkov, Y. and Ramalingam, S.S. (2018) Osimertinib in untreated EGFR-mutated advanced non-small-cell lung cancer. N. Engl. J. Med., 378(2), 113–125.
- US FDA approves Tagrisso as 1st-line treatment for EGFR-mutated non-small cell lung cancer (AstraZeneca press release, 18 April 2018).
- Forget Tagrisso's $3B sales target. It'll be worth double that by 2023, analyst says (FiercePharma press release, 25 June 2018).
1st September 2018, Tafenoquine
Five species of parasitic single-celled microorganisms of the Plasmodium genus are known to cause malaria in humans. P. falciparum is by far the deadliest, being responsible for more than 90% of global malaria deaths, so has understandably received the lion's share of attention (and funding) in the fight against the disease. In contrast, P. vivax has received much less publicity, but it is responsible for around 8.5 million infections annually. This includes 70–90% of malaria cases throughout most of Asia and South America, 50–60% of cases in Southeast Asia and the Western Pacific, and up to 10% of cases in Africa [1].Vivax malaria used to be officially known as "benign" malaria (in contrast to the "malignant" variety caused by P. falciparum). Thanks to the work of Austrian psychiatrist Julius Wagner-Jauregg (1857–1940), from 1917 until the advent of penicillin antibiotics in the 1940s, it was widely used as a treatment for tertiary syphilis (the end-phase of the disease in which the Treponema pallidum bacteria attacks the brain and nervous system).
Around half of the patients treated with P. vivax recovered enough to resume normal activities. Although it is still not known exactly how the treatment worked, it seems that the prolonged high fevers it induced helped the patients' immune systems to fight off the bacteria. Wagner-Jauregg was awarded the 1927 Nobel Prize in Physiology or Medicine for his discovery (although in later years his reputation suffered through his support for eugenics and forced sterilisation of the mentally ill and criminal, as well as his sympathies for the Nazi party).
The term "benign malaria" is no longer used, for the reality is that vivax malaria is anything but benign. Just like falciparum malaria, it causes the classic malaria symptoms of alternating fever, chills and sweats along with headaches and muscle pain. While P. falciparum attacks red blood cells of all ages, P. vivax targets reticulocytes (immature red blood cells that make up roughly 1% of the red blood cells in the body). This can make vivax infections difficult to diagnose. Furthermore, unlike P. falciparum, P. vivax can populate the bloodstream with sexual-stage parasites (the form carried by mosquitoes on their way to the next victim) before any symptoms of malaria appear, making an outbreak of the disease particularly difficult to stop.
While an initial P. vivax infection can appear to be cured using anti-malarial drugs, the parasites can get into the liver and survive there in a dormant form known as a hypnozoite [2], causing no symptoms and undetectable by blood tests. It is this form that enables P. vivax to survive in more temperate zones, where mosquitoes bite only part of the year. Weeks or months later, the parasite re-emerges and the disease flares up again. Commonly recurring 6-8 times a year, each bout of the disease causes sufferers to become gradually more anaemic.
Fighting off each flare-up of the disease makes sufferers more vulnerable to other diseases (as well as to problems resulting from malnutrition and poverty – they are unable to work while they are ill). In turn, other diseases, including falciparum malaria, seem to trigger relapses of vivax malaria. Vivax malaria infection is consequently a significant factor in many deaths [1].
Until recently, primaquine was the only treatment available to clear the parasites hiding in the liver and so prevent the relapse of vivax malaria. However, it needs to be taken for 14 days and common side effects include nausea, stomach pain, and vomiting. Consequently, many patients who feel better after just a few days stop taking the pills, allowing the parasite to awaken at a later date.
At the Walter Reed Army Institute of Research, a compound synthesised in 1978 which was coded as WR 238,605, later named etaquine but which now has the International Nonproprietary Name tafenoquine (CHEBI:135752) was investigated as a radical cure against relapsing vivax malaria, but for many years the development work was not given high priority [3]. In the last 30 years, however, tafenoquine has been the subject of more than 30 clinical trials and found to be effective against the major malaria parasites at all stages of the parasites' life cycle and to have very little potential to trigger the development of resistance.
In July, the United States Food and Drug Administration (FDA) approved the use of tafenoquine for the radical cure of vivax malaria in patients aged 16 and older [4]. In contrast to primaquine, a single 300 mg dose is all that is required to clear P. vivax from the body. It is the first new treatment for vivax malaria to be approved in over 60 years.
The background image is a detail of a Creative Commons licensed thin film micrograph showing a red blood cell containing two ring-form P. vivax parasites. P. vivax rings have a large quantity of cytoplasm and a large chromatin dot, as well as occasional pseudopods. The red blood cells are normal, to 1.5Å normal, sized, round, and contain fine Schüffner's dots, and quite often contain multiple parasites. (Photograph: CDC/Dr Mae Melvin) From: Icke, G., Davis, R. and McConnell, W. (2005) Teaching health workers malaria diagnosis. PLoS Med., 2(2), e11.
Reference(s)
- Vogel, G. (2013) The forgotten malaria. Science, 342(6159), 684–687.
- Markus, M.B. (2011) Malaria: origin of the term "hypnozoite" J. Hist. Biol., 44(4), 781–786.
- Peters, W. (1999) The evolution of tafenoquine—antimalarial for a new millennium? J. R. Soc. Med., 92(7), 345–352.
- US FDA approves Krintafel (tafenoquine) for the radical cure of P. vivax malaria. (GSK press release, 20 July 2018).
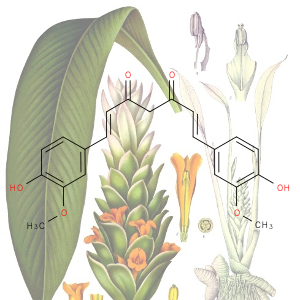
1st August 2018, Curcumin
In a recent paper published by in the Proceedings of the National Academy of Sciences of the U.S.A. [1], a team from the University of California, San Diego, School of Medicine, in collaboration with researchers at Peking University and Zhejiang University, have cast some light on previous reports of the anti-cancer activity of curcumin (CHEBI:3962), the active ingredient in turmeric (Curcuma longa) [2]. The researchers used x-ray crystallography and kinase-inhibitor specificity profiling to show that curcumin binds to a kinase enzyme known as dual-specificity tyrosine-regulated kinase 2 (DYRK2). This binding inhibits the activity of the enzyme, reducing the DYRK2-mediated phosphorylation of the 26S proteasome (a large multi-protein complex involved in the regulated degradation of unwanted or damaged proteins in cells), leading to reduced proteasome activity and impaired cell proliferation.The prime targets for curcumin that give rise to its anti-cancer effects had previously been thought to be the kinase enzymes GSK3 and IKK, but the researchers found that curcumin is over 500 times more potent against DYRK2 (IC50 5 nM) than against GSK3 (IC50 > 3 μM) or IKK (IC50 > 10 μM).
The team found that when curcumin was used in combination with the proteasome inhibitor carfilzomib against the aggressive triple-negative breast cancer (TBNC) cell line, there was a strong synergistic effect – curcumin induced a much higher degree of cancer cell death. An added bonus was that this combination treatment showed little or no cytotoxicity to non-cancerous cells. Use of mouse cancer models also confirmed that curcumin is a selective DYRK2 inhibitor and that DYRK2 is a target with anticancer potential.
While curcumin has demonstrated the "proof of concept", it is unlikely be the final agent of choice. For in a recent review of the medicinal chemistry of curcumin [3], researchers led by Prof. Kathryn Nelson at the Institute for Therapeutics Discovery & Development at the University of Minnesota pointed out that curcumin is a member of the classes known as PAINS (pan-assay interference compounds) [4] and IMPS (invalid metabolic panaceas) [5]. In vivo, curcumin is remarkably unstable (T1/2 < 5 min.) – the review authors liken it to "a missile that continually blows up on the launch pad, never reaching the atmosphere or its intended target". Despite there being more than 15,000 papers published related to biological interactions of curcumin (and typically around 50 more published each week) leading to 120 clinical trials, no double-blind placebo controlled trial of curcuminoids has yet been successful.
The background image is a Creative Commons licensed picture showing different parts of the plant Curcuma longa, published in Köhler's Medicinal Plants.
Reference(s)
- Banerjee, S., Ji, C, Mayfield, J.E., Goel, A., Xiao, J., Dixon, J.E. and Guo, X. (2018) Ancient drug curcumin impedes 26S proteasome activity by direct inhibition of dual-specificity tyrosine-regulated kinase 2. Proc. Natl. Acad. Sci. U. S. A, July 9, 201806797, doi: 10.1073/pnas.1806797115. [Epub ahead of print].
- Wilken, R., Veena, M.S., Wang, M.B. and Srivatsan, E.S. (2011) Curcumin: a review of anti-cancer properties and therapeutic activity in head and neck squamous cell carcinoma. Mol. Cancer, 10, 12.
- Nelson, K.M., Dahlin, J.L., Bisson, J., Graham, J., Pauli, G.F. and Walters, M.A.(2017). The essential medicinal chemistry of curcumin. J. Med. Chem., 60(5), 1620–1637.
- Baell, J. and Walters, M.A. (2014) Chemistry: chemical con artists foil drug discovery. Nature, 513(7519), 481–483.
- Burgos-Morón, E., Calderón-Montaño, J.M., Salvador, J., Robles, A. and López-Lázaro, M. (2010) The dark side of curcumin. Int. J. Cancer , 126(7), 1771–1775.
1st July 2018, Cisplatin
The square planar platinum complex known today as cisplatin (CHEBI:27899, also known as cis-diamminedichloridoplatinum(II)) is historically known as Peyrone's salt, after the Italian chemist Michele Peyrone (1813–1883) who first synthesised it in 1844 [1]. Its structure was finally established some five decades later, as a result of the studies of Swiss chemist Alfred Werner (1866–1919) into the isomerisation of inorganic complexes [2].It was the research of Professor Barnett Rosenberg (1926–2009) and co-workers that would finally raise the status of cisplatin to chemical superstar. In the mid-1960s, they found that the complex prevented bacteria from dividing normally [3]. Subsequent testing on cancer cells showed that it was biologically active. After undergoing animal and clinical trials, cisplatin was approved for treatment of testicular and ovarian cancers in the late 1970s [4].
Cisplatin is administered as an intravenous injection – it's ineffective when given orally – and enters into cancer cells from the bloodstream. Inside the cell, the concentration of chloride ions is much lower than outside, favouring the dissociation of one of the chlorides of the cisplatin and its replacement by water, giving the more reactive complex cis-[PtCl(NH3)2(OH2)]+. In turn, the water can be replaced by one of the N-heterocyclic bases on DNA, particularly guanine. Cross-linking to the DNA can then occur through displacement of the remaining chloride by another guanine group, interfering with mitosis. The cancer cells recognise something is wrong and initiate apoptosis (programmed cell death) [5].
The introduction of cisplatin has had dramatic effects on the success of cancer treatments – before its introduction, the cure rate of testicular cancer was around 10%; today, it is close to 100%. Even though five other platinum drugs with structures based on that of cisplatin have been developed since, cisplatin is still one of the most widely used anticancer agents. It is included in the WHO's Model List of Essential Medicines.
However, cisplatin is by no means a perfect drug. Its use is associated with a number of particularly unpleasant side-effects. The best known are probably severe nausea and vomiting, which are so bad that development work was nearly stopped when the drug was first tested on people; it was only approved following the invention of effective anti-nausea drugs [6]. Other side effects of cisplatin include nephrotoxicity (kidney damage), electrolyte disturbance, including hypomagnesaemia and hypokalaemia, and ototoxicity (hearing loss).
As well as killing cancer cells, cisplatin (and the other platinum-containing drugs) also kills cells in the inner ear, causing permanent damage to the cochlea (the part of the inner ear responsible for hearing) in 40-80% of adults and at least 50% of children. The damage particularly affects hearing high frequency sounds, so can be devastating for young children who have not developed speech, – consonants such as 'f', 'h', and 's' are heard at high frequencies and even mild hearing damage is likely to affect their speech development, learning, and general quality of life.
It has been known since the 1980s that sodium thiosulfate (Na2S2O3, STS) can reduce the cytotoxicity and nephrotoxicity of cisplatin [7]. Subsequent nonclinical studies and phase 1 and phase 2 trials have shown that STS has a potential otoprotective effect [8,9]. It inactivates platinum complexes by binding the electophilic platinum with thiol, forming a non-cytotoxic covalent complex that is rapidly excreted.
Last year, a group of researchers suggested a reason for cisplatin's ototoxicity: they found that in both mice and humans, while cisplatin is eliminated from most organs in the body within a few days or weeks, it is retained in (and damages) the cochlea for months or even years after the treatment [10].
Now, a multi-national team led by Penelope Brock, paediatric consultant at the Great Ormond Street Hospital in London, has reported on a 7-year study of 109 children suffering from hepatoblastoma, a rare liver cancer (1.3 patients per million population) that occurs in infants and children, but is most often diagnosed in children under two [11]. The study group was enrolled at 52 centres in 12 countries from 2007. The children were either given the standard treatment involving cisplatin chemotherapy, or cisplatin followed by STS six hours later (giving the cisplatin some time to work before being deactivated the the STS).
Of the 46 'cisplatin alone' patients, 29 (63%) suffered hearing loss, compared with only 18 of the 55 (33%) of those who were also given STS – corresponding to a 48% reduction in risk. Importantly, the six hour delay between administering the cisplatin and the STS treatment was long enough for the cisplatin to work – the new treatment was just as effective as the traditional 'cisplatin alone' treatment. The 3-year event-free survival rate was 79% in the 'cisplatin alone' group compared with 82% in the 'cisplatin followed by STS' group, while the overall survival rate was 92.3% for the 'cisplatin alone' group and 98.2% for the 'cisplatin followed by STS' group [11].
Almost 40 years after the ototoxicity of cisplatin was discovered, it seems that a big step forward has been made. Sodium thiosulfate (tradename Pedmark) is now being reviewed for a licence for use in hepatoblastoma treatment in Europe and the US. If a license were to be granted in the UK, further review to judge the cost-effectiveness of the treatment would be required before it could be made routinely available. If the work were also shown to be applicable to the treatment of other tumours, it would have a major impact on cancer treatment.
The background image was the first ChEBI logo and depicts a ball and stick model of cisplatin. It was used from the first release of ChEBI in July 2004 until March 2013 (release 99), when it was replaced by the current (caffeine) logo.
Reference(s)
- Peyrone, M. (1844) Ueber die Einwirkung des Ammoniaks auf Platinchlorür. Ann. Chem. Pharm., 51, 1.
- Kauffman, G.B. (1997) Alfred Werner's Research on the Platinum Metals. Platinum Met. Rev., 41(1), 34–40.
- Thomson, A. J. (2007). Christie, D. A.; Tansey, E. M., eds. "The Discovery, Use and Impact of Platinum Salts as Chemotherapy Agent for Cancer". Wellcome Trust Witnesses to Twentieth Century Medicine. 30: 6–15. ISBN: 978-0-85484-112-7.
- Alderden, R.A., Hall, M.D. and Hambley, T.W. (2006) The discovery and development of cisplatin J. Chem. Educ., 83(5), 728–734.
- Wang, D. and Lippard, S.J. (2005) Cellular processing of platinum anticancer drugs. Nat. Rev. Drug Discovery , 4(4), 307–320.
- Wheate, N. and Apps, M. (2015) Happy 50th anniversary to cisplatin, the drug that changed cancer treatment. The Conversation, March 10, 2015.
- Abe., R., Akiyoshi, T., Tsuji, H. and Baba, J. (1986) Protection of antiproliferative effect of cis-diamminedichloroplatinum(II) by sodium thiosulfate. Cancer Chemother. Pharmacol., 18(2), 98–100.
- Neuwelt, E.A., Pagel, M.A., Kraemer, D.F., Peterson, D.R. and Muldoon, L.L. (1984) Bone marrow chemoprotection without compromise of chemotherapy efficacy in a rat brain tumor model. J. Pharmacol. Exp. Ther., 309(2), 594–599.
- Muldoon, L.L., Pagel, M.A., Kroll, R.A., Brummett, R.E., Doolittle, N.D., Zuhowski, E.G., Egorin, M.J. and Neuwelt, E.A. (2000) Delayed administration of sodium thiosulfate in animal models reduces platinum ototoxicity without reduction of antitumor activity. Clin. Cancer Res., 6(1), 309–315.
- Breglio, A.M., Rusheen, A.E., Shide, E.D., Fernandez, K.A., Spielbauer, K.K., McLachlin, K M., Hall, M.D., Amable, L. and Cunningham, L.L. (2017) Cisplatin is retained in the cochlea indefinitely following chemotherapy. Nat. Commun., 8(1), 1654.
- Brock, P.R., Maibach, R., Childs, M., Rajput, K., Roebuck, D., Sullivan, M.J., Laithier, V., Ronghe, M., Dall'Igna., P., Hiyama, E., Brichard, B., Skeen., J., Mateos, M.E., Capra, M., Rangaswami, A.A., Ansari, M., Rechnitzer, C., Veal, G.J., Covezzoli, A., Brugières, L., Perilongo, G., Czauderna, P., Morland, B. and Neuwelt, E.A. (2018) Sodium thiosulfate for protection from cisplatin-induced hearing loss. N. Engl. J. Med., 378(25), 2376–2385.
1st June 2018, WAY-316606
Cyclosporin A (CsA), a metabolite of the fungus Tolypocladium inflatum, has been used since the 1980s as a immunosuppressant for the treatment of autoimmune diseases such as rheumatoid arthritis, psoriasis, Crohn's disease, and nephrotic syndrome, and is also used to prevent tissue rejection following organ transplants. It is included in the WHO's Model List of Essential Medicines.A very common side effect of treatment with CsA is hypertrichosis, a condition in which excessive hair growth can occur anywhere on a person's body. This can occasionally be particularly marked, with some patients experiencing adverse social problems as a consequence [1-3]. CsA is also known to prolong the period of active hair growth in organ-cultured human scalp hair follicles (HFs) ex vivo [4].
It is not surprising, therefore, that a group of researchers trying to find new treatments for hair loss disorders (HLD) should turn their attention to CsA. Although HLD often result in serious psychological problems, only two compounds have so far been approved for treating the condition – minoxidil and finasteride – neither of which is particularly effective [5].
The researchers, led by Dr. Nathan Hawkshaw in the laboratory of Prof. Ralf Paus at the University of Manchester's Centre for Dermatology Research, treated organ-cultured human scalp hair follicles with CsA, then carried out a gene expression profiling of the follicles. They found that the CsA treatment had inhibited the production of secreted frizzled-related protein 1 (SFRP1), a protein that inhibits the growth of hair follicles (and other tissues). This explains why CsA causes unwanted hair growth in patients undergoing CsA treatment [6].
Although CsA has too many toxicity problems to be used for treating hair loss disorders, other SFRP1 inhibitors might be suitable. One such compound is WAY-316606 (CHEBI:140700), which was identified from screening a library of over 440,000 drug-like compounds by American researchers who were searching for a drug for the treatment of osteoporosis [7]. When the Manchester team treated hair follicles with WAY-316606, they found that hair growth was significantly increased [6].
The publication of their results led to major press coverage around the world, some of which optimistically suggested that future treatment of baldness may not be far away (no human trials have yet taken place!). While future steps may include clinical trials, the team's most important discovery is likely to be the potential of the SFRP1 mechanism as a target for new treatments of HLD.
The background image is a detail of a Creative Commons licensed image of gonks (particularly hirsute stuffed toys that were particularly popular in the UK in the 1960s and 1970).
Reference(s)
- Wysocki, G.P. and Daley, T.D. (1987) Hypertrichosis in patients receiving cyclosporine therapy. Clin. Exp. Dermatol., 12(3), 191–196.
- Sternthal, M.B., Murphy, S.J., George, J., Kornbluth, A., Lichtiger, S. and Present, D.H. (2008) Adverse events associated with the use of cyclosporine in patients with inflammatory bowel disease. Am. J. Gastroenterol., 103(4), 937–943.
- Panicker, V.V., Mathew, A. and Dhamramaratnam, A. (2012) Cosmetically disfiguring side effects of cyclosporine. Int. J. Trichology, 4(1), 50.
- Taylor, M., Ashcroft, A.T.T. and Messenger, A.G. (1993) Cyclosporin A prolongs human hair growth in vitro. J. Invest. Dermatol., 100, 237–239.
- Paus, R. (2006) Therapeutic strategies for treating hair loss. Drug Discovery Today: Ther. Strategies, 3(1), 101–110.
- Hawkshaw, N.J., Hardman, J.A., Haslam, I.S., Shahmalak, A., Gilhar, A., Lim, X. and Paus, R. (2018) Identifying novel strategies for treating human hair loss disorders: Cyclosporine A suppresses the Wnt inhibitor, SFRP1, in the dermal papilla of human scalp hair follicles. PLoS Biol., 16(5), e2003705.
- Bodine, P.V.N., Stauffer, B., Ponce-de-Leon, H., Bhat, R.A., Mangine, A., Seestaller-Wehr, L.M., Moran, R.A., Billiard, J., Fukayama, S., Komm, B.S., Pitts, K., Krishnamurthy, G., Gopalsamy, A., Shi, M., Kern, J.C., Commons, T.J., Woodworth, R.P., Wilson, M.A., Welmaker, G.S., Trybulski, E.J. and Moore, W.J. (2009) A small molecule inhibitor of the Wnt antagonist secreted frizzled-related protein-1 stimulates bone formation. Bone, 44(6), 1063–1068..
1st May 2018, Valproic acid
Valproic acid (CHEBI:39867, also known as VPA, dipropylacetic acid, DPA, 2-propylvaleric acid, or 2-propylpentanoic acid) was first synthesised as an analogue of the valerian metabolite valeric acid (pentanoic acid) in 1881 [1]. A high-boiling (220 °C), colourless liquid that is very soluble in organic solvents but almost insoluble in water, it became widely used as a solvent for organic chemicals. However, its remarkable anticonvulsant (antiepileptic) properties only came to light by chance some 80 years after it was first synthesised. As part of his thesis in 1962, Pierre Eymard, working under the supervision of G. Carraz at the School of Medicine and Pharmacy in Grenoble, France, had synthesised several derivatives of khelline. Two colleagues, H. Meunier and Y. Meunier, working for Berthier Laboratories in Grenoble, had used VPA for dissolving a bismuth salt. So Eymard, Meunier and Meunier decided to dissolve some of the khelline derivatives in VPA and test their pharmacological activities. Carraz suggested using the pentylenetetrazole (PTZ) seizure test for antiepileptic activity. All of the compounds tested seemed to prevent PTZ-induced convulsions in laboratory rats – a remarkable result for routine screening tests [2,3].It soon became apparent that it was the solvent rather than the test compounds that possessed the anticonvulsant properties. Clinical trials had started by 1964 and VPA (normally used as its sodium or semisodium salts; the term 'valproate' is used to mean both valproic acid and its salts) was first approved for use as an antiepileptic drug in France in 1967. Fifty years on, valproate is the most widely prescribed antiepileptic drug in the world and is included in the WHO's Model List of Essential Medicines. In addition to treating and preventing epileptic seizures, it is also used in the treatment of bipolar disorder and for the prevention of migraines.
Pregnant women who suffer from epilepsy have long been known to have higher rates of pre-term delivery, low birth weight, still-births, neonatal losses, and maternal deaths than in the general population. An increased risk of fetal malformation has also been observed. The causes are complex, and can include the disease itself and the number of seizures suffered during pregnancy. In addition, all currently used antiepileptics are now known to have teratogenic effects [4].
A rare condition known as fetal valproate syndrome was first reported over 30 years ago. Children born to women who took valproate during their pregnancy have been found to be at significantly greater risk of suffering autism, heart defects, spina bifida, urogenital defects and orofacial clefts than in the general population [5,6]. It is thought that in the UK alone, up to 20,000 children have been born with disabilities caused by valproate since the drug was first approved in the 1970s. Even more worryingly, a 2016 study on mice suggests that symptoms of autism may subsequently be inherited by the next generation [7].
In the UK, the information provided to patients prescribed valproate has included a warning about the possible risk of birth defects since the drug was first approved in 1974. As understanding of the extent of the risks of taking valproate in pregnancy improved (it is now estimated that up to 4 in 10 babies are at risk of developmental disorders and around 1 in 10 are at risk of birth defects), so the warnings to patients taking the drug were strengthened. Even so, a 2017 survey commissioned by the three main UK epilepsy charities found that one in five women prescribed sodium valproate were unaware that taking it during pregnancy could harm the unborn baby [8].
Now, the UK Medicines and Healthcare products Regulatory Agency (MHRA), which is responsible for ensuring medicines work and are acceptably safe, has changed the license for valproate. Medicines containing valproate cannot be prescribed to women or girls of childbearing age unless a "Pregnancy Prevention Programme" is in place. This involves an initial negative pregnancy test confirmed by a healthcare professional, and an undertaking to use highly effective contraception. Their treatment must be reviewed at least annually, and at each review the woman must sign a risk acknowledgement form [9].
The background image is a Creative Commons licensed image of generalised 3 Hz spike and wave discharges in a child with childhood absence epilepsy.
Reference(s)
- Burton, B.S. (1882) On the propyl derivatives and decomposition products of ethyl acetoacetate. Am. Chem. J., 3(6), 385–395.
- Scott, D.F. (1993) The History of Epileptic Therapy: An Account of How Medication was Developed 131–132 (CRC Press).
- Löscher, W. (2012) The discovery of valproate. In Valproate (ed. Löscher, W.) 1–3.
- Güveli, B.T., Rosti, R.Ö., Güzeltaș, A, Tuna E.B., Atakli, D., Sencer, S., Yekeler, E., Kayserili, H., Dirican, A., Bebek, N., Baykan, B., Gökyiğit, A. and Gürses, C. (2017) Teratogenicity of antiepileptic drugs. Clin. Psychopharmacol. Neurosci., 15(1), 19–27.
- Clayton-Smith, J. and Donnai, D. (1995) Fetal valproate syndrome. J. Med. Genet., 32(9), 724–727.
- Mohd Yunos, H. and Green, A. (2018) Fetal valproate syndrome: the Irish experience. Ir. J. Med. Sci., Feb. 3. doi: 10.1007/s11845-018-1757-6.
- Choi, C.S., Gonzales, E.L., Kim, K.C., Yang, S.M., Kim, J.W., Mabunga, D.F., Cheong, J.H., Han, S.H., Bahn, G.H. and Shin, C.Y. (2016) The transgenerational inheritance of autism-like phenotypes in mice exposed to valproic acid during pregnancy. Sci. Rep., 6, 36250.
- Hutchinson, S. (2017) Epilepsy drug warnings 'not reaching women', survey shows. BBC News, 22 Sept. 2017.
- Valproate banned without the pregnancy prevention programme (Medicines and Healthcare products Regulatory Agency press release), 24 April 2018.
1st April 2018, Pavinetant
Menopause, the permanent cessation of menstruation and reproductive capability caused by reduced estrogen levels, is one of the most significant events in a woman's life resulting in several permanent physiological changes. The average menopause lasts for seven years, and approximately four in five women will experience menopausal symptoms, such as hot flushes, sleep disruption, poor concentration and depression. In numerous cases, these symptoms can be severe and have an adverse effect on quality of life. Previously, women have been treated with hormone replacement therapy (HRT), which contains estrogen, but this treatment has been linked to an increased risk of breast and ovarian cancers as well as blood clots. Now, a team of researchers led by Professor Waljit Dhillo at Imperial College London have found a new class of experimental drugs that reduce hot flushes in menopausal women by almost three-quarters in a relatively short treatment time. The research, funded by the Medical Research Council and the National Institute for Health Research, is a new in-depth analysis of data collected from a clinical trial initially published in 2017 [1]. The initial study comprised a randomised double-blind placebo-controlled clinical trial, involving 37 menopausal women aged between 40 and 62 years old who were experiencing seven or more hot flushes a day. Participants were randomly chosen to first receive either an 80 mg daily dose of a drug, called pavinetant (CHEBI:140478, also known as MLE4901), or a placebo over the course of a four-week period, before the groups were switched. The researchers found that the drug significantly reduced both the total number and severity of flushes during the four-week treatment period, compared to the patients who received the placebo over the same period.In a more in-depth analysis of the data, the team were able to demonstrate that taking MLE4901 for just three days had significant beneficial effects [2]. The new data showed that the drug reduced the number of hot flushes by almost three-quarters and also decreased their severity. Furthermore, the women reported a 82% decrease in the amount their hot flushes disrupted sleep patterns, and a 77% reduction in interruption to their concentration all within the three-day window. It is thought that MLE4901 works by blocking neurokinin-3 (NK3) receptors in the brain, thus preventing NK3 from activating temperature control areas and triggering hot flushes. The drug, originally developed as a potential drug for schizophrenia, will not be taken any further in clinical trials due to adverse side effects that affect liver function. However, two structural analogues of MLE4901, which also block NK3 receptors but do not appear to exhibit any side effects are currently undergoing larger patient trials. The team hope that this new class of drugs will ultimately provide a safer alternative to HRT.
Our background image is a creative commons licensed image depicting a glass bottle used for Progynon pills, the first orally active formulation of estrogen to be introduced for medical use.
Reference(s)
- Prague, J.K., Roberts, R.E., Comninos, A.N., Clarke, S., Jayasena, C.N., Nash, Z., Doyle, C., Papadopoulou, D.A.,Bloom, S.R., Mohideen, P., Panay, N., Hunter, M.S., Veldhuis, J.D., Webber, L.C., Huson, L. and Dhillo, W.S. (2017). Neurokinin 3 receptor antagonism as a novel treatment for menopausal hot flushes: a phase 2, randomised, double-blind, placebo-controlled trial. Lancet, 389, 1809–1820.
- Prague, J.K., Roberts, R.E., Comninos, A.N., Clarke, S., Jayasena, C.N., Mohideen, P., Lin, V.H., Stern, T.P., Panay, N., Hunter, M.S., Webber, L.C. and Dhillo, W.S. (2018). Neurokinin 3 receptor antagonism rapidly improves vasomotor symptoms with sustained duration of action. Menopause, doi:10.1097/GME.0000000000001090, published online 12 March 2018.
1st March 2018, L-asparagine
The first amino acid ever isolated, L-asparagine (commonly known just as asparagine and abbreviated to L-Asn or N, CHEBI:17196) gets its name from the asparagus (Asparagus officinalis) juice from which it was first isolated over 200 years ago by the French chemists Louis Nicolas Vauquelin (1763–1829) and Pierre Jean Robiquet (1780–1840) [1]. One of the 20 standard proteinogenic amino acids, it is synthesised in the body from oxaloacetic acid via L-aspartic acid. Since it does not have to be supplied in the diet, it is classed as a non-essential amino acid. In recent years, asparagus has been frequently described as a 'superfood', with its high asparagine content being included among its supposed benefits. In reality, the term 'superfoods' is seldom used by dietitians or nutrition scientists, and usually has more to do with marketing than any genuine benefits [2]. In a recent paper in Nature, however, a team of cancer specialists from the UK, the USA, and Canada report that, despite the purported nutritional benefits, asparagine in the diet could be making things worse for people suffering from breast cancer [3].The main cause of death among breast cancer patients is not the primary tumour, but secondary tumours, known as metastases, secondary tumours have already spread (from the primary tumour) by definition. They can appear anywhere, but often do so in the brain, lungs, or bones. The researchers studied a group of mice with a particularly aggressive strain of breast cancer – untreated mice tended to develop secondary tumours in weeks and died within a few months. The team studied the effect of reducing the bioavailability of asparagine to the tumours using three different methods: by blocking asparagine synthetase (the key enzyme that produces asparagine), by treating the mice with L-asparaginase (an enzyme that converts asparagine to aspartic acid and ammonia), or by restricting the amount of asparagine in the diet.
The investigators found that while the treatments did not prevent primary tumours from forming, they significantly reduced their metastatic spread around the body. In contrast, giving the mice high-asparagine diets increased the metastatic spread. It appears as though asparagine has an effect on proteins in the cancer cells that promote epithelial-to-mesenchymal transition, a process in which the cancer cells change into a form that easily spreads through the bloodstream to other organs, where they grow into secondary tumours.
If the group's results are also found to apply to humans, then it is possible that breast cancer patients may in future be put on 'low-asparagine' diets. However, since asparagine occurs very widely in foods – animal sources include beef, poultry, eggs, fish and seafood, while plant sources include potatoes, legumes, nuts, and soya – drug treatments aimed at blocking the production of asparagine and increasing its breakdown in the body would probably be more effective.
When the team examined records from human cancers, they found that breast tumours that produced the most asparagine were the most likely to spread, causing patients to die sooner. Similar results were seen in cancers of the head, neck and kidney. So while breast cancer is the most common cancer in the UK, where it accounts for 15% of all new cancer cases and kills almost 1,000 women each month, the findings could also be relevant for the treatment of other human cancers.
The background image is a Creative Commons licensed photograph of pink, blue and white silage bales at Gåseberg Sheep Farm, Lysekil Municipality, Sweden. The pink silage wrapping is sold to support breast cancer awareness and the blue for prostate cancer awareness. Part of the proceeds from the sale of the wrappings goes to the respective cancer research. The white bales are the normal colour.
Reference(s)
- Vauquelin, L.N. and Robiquet, P.J. (1806) La découverte d'un nouveau principe végétal dans le suc des asperges". Ann. Chim., 57, 88–93.
- Cancer Research UK. Food Controversies: Superfoods.
- Knott, S.R.V., Wagenblast, E., Khan, S., Kim, S.Y., Soto, M., Wagner, M., Turgeon, M.O., Fish, L., Erard, N., Gable, A.L., Maceli, A.R., Dickopf, S., Papachristou, E.K., D'Santos, C.S., Carey, L.A., Wilkinson, J.E., Harrell, J.C., Perou, C.M., Goodarzi, H., Poulogiannis, G. and Hannon, G.J. (2018) Asparagine bioavailability governs metastasis in a model of breast cancer. Nature, 554(7692), 378–381.
1st February 2018, Triclosan
Malaria is known to cause around half a million deaths per year, predominantly in Africa and south-east Asia where mosquitos are in abundance. Approximately 90% of these deaths are caused by Plasmodium falciparum, the parasite responsible for the most severe forms of the disease. While a number of medicines are used to treat the disease, malaria parasites are becoming increasingly resistant to these drugs raising the risk that some strains may eventually become untreatable. In a recent study, a team of researchers at Cambridge University have discovered that triclosan (CHEBI:164200), an ingredient found in soaps, deodorants and some toothpastes, is active against strains of malaria parasites that have grown resistant to established antimalarial drugs [1]. The team led by Dr. Elizabeth Bilsland and Professor Steve Oliver were assisted by an artificially-intelligent (AI) robot nicknamed "Eve".After being transferred into a human host via a mosquito bite, malaria parasites work their way into the liver, where they mature and reproduce. After a few days, the parasites leave the liver and hijack red blood cells, where they continue to multiply, spreading around the body and causing potentially life-threatening symptoms. When used in toothpaste, triclosan prevents the build-up of plaque bacteria by inhibiting the action of enoyl reductase (ENR), an enzyme found in the liver and involved in the fatty acid synthesis (FAS) pathway. It has been known for some time that triclosan also inhibits the growth of the P. falciparum during the blood stage, and the assumption was that the drug was targeting ENR [2]. However, subsequent work proved convincingly that the FAS pathway was not an important factor in the proliferation of P. falciparum in the blood [3]. Hence action against a different target must be responsible for triclosanâs inhibition of the growth of blood stage P. falciparum.
An AI robot known as 'Eve', developed by teams of scientists based at the Universities of Manchester, Aberystwyth, and Cambridge with the specific purpose of speeding up the drug discovery process, was used by the Cambridge team to perform automated high-throughput screening of an array of FDA-approved compounds and discovered that in fact, triclosan disrupts proliferation of P. falciparum by specifically inhibiting a different enzyme, called dihydrofolate reductase (DHFR), also a target of established antimalarial drugs such pyrimethamine. Next, the team demonstrated that triclosan was able to target and act on DHFR even in pyrimethamine-resistant strains of P. falciparum. The ability to target two points in the lifecycle of the malaria parasite should make it difficult for the parasite to evolve resistance.
This example of collaboration between man and machine has provided what could be a valuable lead compound in the development of a novel class of antimalarial drugs.
Our background image is a creative commons licensed image depicting a tube of toothpaste.
Reference(s)
- Bilsland, E., van Vliet, L., Williams, K., Feltham, J., Carrasco, M.P., Fotoran, W.L., Cubillos, E.F.G., Wunderlich, G., Grøtli, M., Hollfelder, F., Jackson, V., King, R.D. and Oliver, S.G. (2018). Plasmodium dihydrofolate reductase is a second enzyme target for the antimalarial action of triclosan.Sci. Rep., doi:10.1038/s41598-018-19549-x, published online 18 January 2018.
- Surolia, N. and Surolia, A. (2001). Triclosan offers protection against blood stages of malaria by inhibiting enoyl-ACP reductase of Plasmodium falciparum. Nat. Med. 7, 167–173.
- Freundlich, J.S., Wang, F., Tsai, H.C., Kuo, M., Shieh, H.M., Anderson, J.W., Nkrumah, L.J., Valderramos, J.C., Yu, M., Kumar, T.R., Valderramos, S.G., Jacobs, W.R., Schiehser, G.A., Jacobus, D.P., Fidock, D.A. and Sacchettini, J.C. (2007).X-ray structural analysis of Plasmodium falciparum enoyl acyl carrier protein reductase as a pathway toward the optimization of triclosan antimalarial efficacy. J. Biol. Chem., 282, 25436–25444.

1st January 2018, Poly(ethylene glycol)
In 1859, when structural organic chemistry was very much in its infancy, two researchers independently described obtaining polymeric products from reactions of ethylene glycol. A. Lourenço heated ethylene glycol with ethylene dibromide in sealed tubes and isolated the products by distillation [1], while Adolphe Wurtz reacted ethylene glycol with ethylene oxide, water, and acetic acid [2].Both researchers isolated a number of polymeric products with the general formula C2nH4n+2On+1, where n is the number of units of the monomer that had been joined to make the polymer molecule. The products are examples of compounds known today as poly(ethylene glycol) (CHEBI:46793). They are also known as polyethylene oxide (PEO) or polyoxyethylene (POE), depending on their molecular weight, but are best known in the biomedical community by the abbreviation PEG.
PEGs are available commercially with average molecular weights ranging from 300 to 10,000,000 (the particular value is often indicated by including it after the name, so PEG-400 has an average molecular weight of 400, equivalent to around nine units of the monomer linked together). As PEGs contain a high proportion of oxygen atoms, regularly spaced along the length of the polymer chain, and as each oxygen can form a hydrogen bond to any water molecule that is in the vicinity, the result is that PEGs have an amazing capacity for binding to water. Furthermore, they are generally colourless, odourless, non-irritating and non-toxic. As a consequence, they have found use in hundreds of diverse applications, including toothpastes, breath fresheners, mouthwashes, skin creams, and printer inks. In most of these applications, PEGs serve to keep the ingredients in solution and increase the shelf-life.
PEGs are also used in many more specialised areas. One of these is archaeology. During the early excavation of the 2,000 year old Terracotta Army in China, archaeologists found that within a few seconds of terracotta pieces being first unearthed and exposed to the dry air, the lacquer under the paint started to curl; within a few minutes, it would flake off. The problem was solved when the Bavarian State Conservation Office developed a PEG-based preservative which, when immediately applied to unearthed artifacts, helped in retaining the ancient colours [3].
If waterlogged wood recovered by marine archaeologists from ancient shipwrecks is allowed to dry naturally, it tends to undergo massive shrinkage, warping and cracking as the water evaporates from the cellular structure of the wood. PEGs are widely used prevent this. One of the best known examples of this followed the recovery of the hull of Henry VIII's flagship, Mary Rose. This was sprayed for 10 years with a low-molecular-weight PEG to replace the water in the cells of the wood, then for a further 7 years with a higher-molecular-weight PEG to seal the outer layers of timber; the PEG-treated hull was then gently air-dried for a further 6 years [4].
In the medical field, probably the best-known use for PEGs is as a hyperosmotic laxative (PEGs bind with water in the stools, preventing it from being reabsorbed by the intestines; the stools remain soft, heavy, and easier to pass). In recent years, however, PEGs have become increasingly used in the creation of protein-based drugs. These come in various types, including recombinant human proteins (e.g. insulin; growth hormone), monoclonal antibodies (used in the treatment of a wide range of diseases, including cancer, haemophilia, rheumatoid arthritis, multiple sclerosis, cardiovascular disease and psoriasis); others are bacterial or viral proteins.
If used in their native form, protein-based drugs can be very short-lived in the body and can trigger inflammation and other immune responses. To get round these problems, the proteins are often modified by PEGylation – that is, by attaching PEG molecules to specific sites on the protein. The PEG groups increase the circulating lifetime of the drug in the body by reducing kidney clearance and by shielding the protein from proteolytic enzymes. The immunogenicity and antigenicity of the protein–PEG complex is also reduced compared with the parent protein. An additional benefit is that the protein–PEG complex tends to be more soluble and easier to handle than the parent protein [5,6].
There are problems with the technique, however. While early difficulties related to specificity (i.e. where the protein is PEGylated) have been largely overcome by the introduction of specific PEGylating reagents, such as monomethoxypoly(ethylene glycol)-maleimide (mPEG-MAL, which is particularly reactive with the thiol group of cysteine residues) [7], a continuing problem has been low reaction yields.
Now a group of researchers based in Beijing have reported that yields for the PEGylation reaction can be dramatically improved by using high pressure conditions [8]. Taking recombinant human ciliary neurotrophic factor (rhCNTH) as their model protein, they found that using mPEG-MAL with an average molecular weight of 40,000 at normal pressure for 5 hours, the PEGylation yield was just 5%. At higher pressures, however, the yield increased dramatically, reaching 90% when the reaction was run at 250 MPa (ca. 2,500 atmospheres). The group also found that, in contrast to the conventional conditions, in which short-chain PEG gives higher yields than long-chain PEG, the high pressure technique afforded similar yields irrespective of the chain length of the mPEG-MAL used.
The team suggest that at normal pressures, a lot of the target amino acids are embedded within the folds of the protein, and so are inaccessible to the PEGylating agent. By increasing the pressure, the protein temporarily unfolds, allowing the PEGylation agent access. If the pressure is increased too much (in this case, beyond 250 MPa) the protein becomes irreversibly denatured. The researchers believe that the high-pressure technique may have potential application in preparing conjugates of other biomacromolecules (although it can only work for proteins that can undergo reversible pressure-induced denaturation).
The background image is a Creative Commons licensed photograph of The Tudor period carrack Mary Rose under going conservation with polyethylene glycol at the Portsmouth Historic Dockyard, United Kingdom.
Reference(s)
- Lourenço, A. (1859) Note sur un éther intermédiaire du glycol. C. R. Hebd. Seances Acad. Sci., 49, 619.
- Wurtz, A. (1859) Synthèse du glycol avec l'oxyde d'éthylène et l'eau. C. R. Hebd. Seances Acad. Sci., 49, 813.
- Larmer, B. (2012) Terra-cotta warriors in color. National Geographic, 221(6), 74–87.
- The Mary Rose Trust. Raising the Mary Rose. Working ashore 1983–.
- Kochendoerfer, G. (2003) Chemical and biological properties of polymer-modified proteins. Expert Opin. Biol. Ther., 3(8), 1253–1261.
- Fee, C. and Damadoran, V.B. (2010) Protein PEGylation: an overview of chemistry and process considerations. Eur. Pharmaceut. Rev., (1).
- Pfister, D. and Morbidelli, M. (2014) Process for protein PEGylation. J. Controlled Release, 180, 134–149.
- Wang, Q., Zhang, C., Guo, F., Li, Z., Liu, Y. and Su, Z. (2017) Novel bioconjugation strategy using elevated hydrostatic pressure: a case study for the site-specific attachment of polyethylene glycol (PEGylation) of recombinant human ciliary neurotrophic factor. Bioconjugate. Chem., 28(11), 2841–2848.
2017
1st December 2017, Bryostatin 1
Bugula neritina is a rather inconspicuous marine organism that resembles seaweed but is actually an invertebrate. It is an abundant and invasive species that is widely viewed as a pest as it accumulates on ships, docksides, buoys and intake valves. It is also the only known source of a very rare group of macrolides known as bryostatins, the most abundant of which, bryostatin 1 (CHEBI:88353) has shown considerable promise in the treatment of cancer, Alzheimer's disease and HIV. However, the longstanding scarcity of the compound has severely limited both research and clinical studies and has hampered efforts to access more potent synthetic analogues. When scientists from the National Cancer Institute (NCI) went searching for bryostatins in 1991, they harvested 14 tons of B. neritina and managed to extract just 18 grams of pure bryostatin 1, equating to a yield of 0.00014%. In addition it was found that the organism only produced bryostatin at depths greater than 10 feet, in warmer seas and only at certain times of the year. Fortunately bryostatin 1 is extremely potent: a single gram is sufficient to treat 1,000 cancer patients or 2,000 Alzheimer's patients. For basic research, only milligram quantities are required.Approaches to solving this supply problem, including the use of supercritical carbon dioxide to extract the compound from its marine source, aquaculture methods and engineered biosynthesis have proved to be either environmentally disruptive or unsustainably expensive. As a result the most viable solution would be to chemically synthesise the compound in the laboratory from scratch. However this is extremely challenging as the macrocyclic lactone structure of bryostatin 1 is highly complex, containing three tetrahydropyran rings, 11 stereocentres and an imposing array of multiple carbon-carbon double bonds, alcohol, ether, hemiketal and ester functionalities.
The first successful total synthesis of bryostatin 1 was reported in 2011, but required 57 synthetic steps and only produced milligram quantities of bryostatin 1 [1]. Now, however a team of researchers led by Paul Wender, a professor of chemistry at Stanford University has reported a convergent 29-step synthesis that produces multigram quantities of bryostatin 1 [2]. Two groups working in parallel devised stereocontrolled and efficient synthetic routes to two fragments of bryostatin 1 and then brought the fragments together via a macrocyclisation reaction. Four further synthetic steps produced 2 grams of bryostatin 1 in an overall yield of 4.8%, tens of thousands of times more efficient that the extraction method. Spectral and analytical techniques proved that that the synthesised product was identical to a natural sample provided by the NCI. What makes the process remarkable is that none of the 29 steps proceeded in a yield less than 75%. It is thought that once production is scaled up, manufacturers could potentially produce about 20 grams of bryostatin 1 per year, sufficient to cover clinical and research requirements.
Our background image is a creative commons licensed picture of the feathery branched Bugula neritina in close proximity to the marine sponges Phorbas tenacior and Dysidea fragilis and the sea-slug Diaphorodoris papillata
Reference(s)
- Keck, G.E., Poudel, Y.B., Cummins, T.J., Rudra, A. and Covel, J.A. (2011) Total synthesis of bryostatin 1. J. Am. Chem. Soc., 133, 744–747.
- Wender, P.A., Hardman, C.T., Ho, S., Jeffreys, M.S., Maclaren, J.K., Quiroz, R.V., Ryckbosch, S.M., Shimizu, A.J., Sloane, J.L. and Stevens, M.C. (2017) Scalable synthesis of bryostatin 1 and analogs, adjuvant leads against latent HIV. Science, 358, 218–223.
1st November 2017, Psilocybin
In our Entity of the Month article for August 2014, we highlighted the discovery by a team of researchers led by Enzo Tagliazucchi at Goethe University Frankfurt that psilocybin (CHEBI:8614), the active ingredient in magic mushrooms, induces changes in the brain that are similar to those that occur during dreams [1]. These observations have led to investigation of the use of hallucinogenic agents as potential antidepressants. Depression is a disabling disease that affects many people. While antidepressants and therapy are effective in many cases, there are numerous instances where people don't fully respond to treatment.In 2016 Robin Carhart-Harris, head of psychedelic research at Imperial College London, and his colleagues conducted the first clinical trial of psilocybin as a means to treat depression [2]. The trial only involved 12 volunteers with treatment-resistant depression and no control group, but results were encouraging. The team observed that depressive symptoms in all 12 patients were markedly reduced 1 week after high-dose treatment with psilocybin.
Now in a follow-up study the same team have demonstrated clear changes in brain activity in depressed people treated with psilocybin after failing to respond to conventional treatments [3]. The study involved 19 volunteers, who were each given a 10 mg dose of psilocybin followed by a 25 mg dose seven days later. The team used functional magnetic resonance imaging to measure changes in blood flow and the crosstalk between brain regions, with patients recording their depressive symptoms via clinical questionnaires. Immediately following treatment, patients reported a decrease in depressive symptoms, such as improvements in mood and stress relief. Brain images of the patients revealed reduced blood flow in some areas of the brain, including the amygdala, a small almond-shaped region of the brain known to be involved in processing emotional responses, stress and fear. These changes in brain activity were associated with a marked and lasting reduction in depressive symptoms with participants in the trial reporting beneficial effects lasting up to five weeks after treatment. Several patients spoke of feeling 'reset' or 'rebooted'. The researchers have acknowledged that the significance of their results is limited by the small sample size and the absence of a placebo group for comparison. They have also warned that that patients with depression should not under any circumstances attempt to self-medicate. The team currently plan to test psilocybin against a leading antidepressant in a more extensive trial set to commence in 2018.
Our background image is a creative commons licensed picture of Psilocybe weilii, a species of magic mushroom having psilocybin as its main active compound.
Reference(s)
- Tagliazucchi, E., Carhart-Harris, R., Leech, R., Nutt, D. and Chialvo, D.R. (2014). Enhanced repertoire of brain dynamical states during the psychedelic experience. Hum. Brain Mapp., 35, 5442–5456.
- Carhart-Harris, R.L., Bolstridge, M., Rucker, J., Day, C.M., Erritzoe, D., Kaelen, M., Bloomfield, M., Rickard, J.A., Forbes, B., Feilding, A., Taylor, D., Pilling, S., Curran, V.H., and Nutt, D.J. (2016). Psilocybin with psychological support for treatment-resistant depression: an open-label feasibility study. Lancet Psych., 3, 619–627.
- Carhart-Harris, R.L., Roseman, L., Bolstridge, M., Demetriou, L., Pannekoek, J.N., Wall, M.B., Tanner, M., Kaelen, M., McGonigle, J., Murphy, K., Leech, R., Curran, H.V. and Nutt, D.J. (2017). Psilocybin for treatment-resistant depression: fMRI-measured brain mechanisms. Sci. Rep., DOI: 10.1038/s41598-017-13282-7, published online 13 October 2017.
1st October 2017, Gemcitabine
The role of serendipity has been a common occurrence throughout the history of scientific innovation and discovery. Examples include Alexander Fleming's accidental discovery of penicillin in 1928 and the invention of the microwave oven by Percy Spencer in 1945. Although early epidemiological studies suggested that coffee consumption could increase the risk of some cancers, thanks to a recent serendipitous discovery, researchers have uncovered one reason why certain chemotherapy drugs are sometimes ineffective. It seems that bacteria present inside cancer cells can metabolise some drugs, rendering them completely inactive. The study was carried out in the laboratory of Dr. Ravid Straussman at the Weizmann Institute of Science in Israel, led by his research student Leore Geller in collaboration with Dr. Todd Golub and Dr. Michal Barzily-Rokni of the Broad Institute of the Massachusetts Institute of Technology [1]. Straussman's team had previously published a study showing how in some cases, the presence of healthy cells in tumours could cause resistance to chemotherapy [2].In the current study, the team were puzzled when they observed that one particular group of skin cells was preventing the chemotherapy drug gemcitabine (CHEBI:175901) from killing neighbouring pancreatic cancer cells. They noticed that the skin cells were infected with Mycoplasma bacteria, but initially dismissed it as external contamination. However, further investigation revealed that the bacteria were actually degrading the drug and causing it to become completely inactive. The researchers then demonstrated that the gene responsible was a "long form" of an enzyme called cytidine deaminase (CDD). In contrast, the corresponding "short form" of CDD could not degrade gemcitabine. Next the group examined over 100 biopsies from human pancreatic cancer patients and found that bacteria producing the long form of CDD were present in the tumours. These included very common bacteria belonging to the class of Gammaproteobacteria, including E. coli and Salmonella spp. Further experiments in mouse models of cancer were carried out using a group of mice infected with bacteria that had the long form CDD gene while a second group were exposed to bacteria lacking CDD. As anticipated, only the group of mice with the CDD gene intact exhibited resistance when the drug was administered. After treatment with antibiotics, this group of mice also responded to the chemotherapy drug.
These initial results are encouraging as pancreatic cancer is a particularly deadly form of the disease because it progresses fast and is usually discovered at an advanced stage. Few patients make a full recovery from it. There are also potential complications as the requirement for antibiotics could result in the generation of antibiotic-resistant bacteria. There is also the possibility of developing drugs that inhibit CDD. The team are currently investigating bacterial disruption of another chemotherapy drug called oxaliplatin.
The background image is a detail from a Creative Commons licensed image depicting Salmonella bacteria growing on XLD agar.
Reference(s)
- Geller, L.T., Barzily-Rokni, M., Danino, T., Jonas, O.H., Shental, N., Nejman, D., Gavert, N., Zwang, Y., Cooper,Z.A., Shee, K., Thaiss, C.A., Reuben, A., Livny, J., Avraham, R., Frederick, D.T., Ligorio, M., Chatman, K., Johnston, S.E., Mosher, C.M., Brandis, A., Fuks, G., Gurbatri, C., Gopalakrishnan, V., Kim, M., Hurd, M.W., Katz, M., Fleming, J., Maitra, A., Smith, D.A., Skalak, M., Bu, J., Michaud, M., Trauger, S.A., Barshack, I., Golan, T., Sandbank, J., Flaherty, K.T., Mandinova, A., Garrett, W.S., Thayer, S.P., Ferrone, C.R,. Huttenhower, C., Bhatia, S.N., Gevers, D., Wargo, J.A., Golub, T.R. and Straussman, R. (2017). Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science (New York, N.Y.), 357, 1156–1160.
- Straussman. R., Morikawa, T., Shee, K., Barzily-Rokni, M., Qian, Z.R., Du, J., Davis, A., Mongare, M.M., Gould, J., Frederick, D.T., Cooper, Z.A., Chapman, P.B., Solit, D.B., Ribas, A., Lo, R.S., Flaherty, K.T., Ogino, S., Wargo, J.A. and Golub, T.R. (2012).Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 487, 500–504.

1st September 2017, Cafestol
Now the most frequently consumed beverage worldwide, coffee is believed to have been introduced into Arabia from Ethiopia during the fifteenth century, reaching Europe by around 1600. In England, the first coffee house opened in Oxford in 1650; by 1675, there were nearly 3,000 [1]. Today, there are around 23,000 and the number is growing by about 6% a year [2]. Although early epidemiological studies suggested that coffee consumption could increase the risk of some cancers, more comprehensive studies now suggest that coffee has neutral or positive effects on human health [3,4].While the stimulant caffeine is probably the best known compound found in coffee, there are well over a thousand others, including alkaloids; phenolic acids such as caffeic, ferulic and quinic acids and their ester-like chlorogenic acid; diterpenoids; lignan; flavonoids; and Maillard reaction products [1]. Most have received relatively little attention from the research community, but a notable exception is cafestol (CHEBI:3291), which was first isolated from coffee oil in 1938 [5,6], although its pentacyclic diterpenoid structure was not determined until 1959 [7,8].
Cafestol has been found to have a number of interesting biological properties, including anti-cancer, pro-apoptotic, and anti-inflammatory activity [9-12]. Not all of its effects are beneficial, however. Investigation into the association of a high intake of Scandinavian-type boiled coffee with hypercholesterolaemia and the risk of coronary heart disease in Norway and Finland [13] eventually identified the culprit as cafestol, which was found to be the most powerful cholesterol-elevating compound in the human diet [14].
In recent years, it has been found that people who drink 3-4 cups of coffee a day have 25% lower relative risk of developing type-2 diabetes (T2D) than those who drink one cup or less [15]. The effect is seen irrespective of whether the coffee drinkers have been drinking caffeinated or decaffeinated coffee – so caffeine is unlikely to be responsible. The antidiabetic effect is also seen irrespective of whether the coffee is filtered or unfiltered. This observation might also seem to rule out cafestol as the bioactive agent, as the amount of cafestol in coffee varies dramatically depending on how the drink is prepared: a cup of Turkish coffee can contain over 4 mg of cafestol, while a cup of filter coffee may contain only 0.1 mg, as virtually all of the diterpenoid is retained on the paper filter [16].
In 2015, however, a group of researchers led by Søren Gregersen at Aarhus University Hospital in Denmark reported that, in in vitro studies, cafestol can increase glucose uptake in human skeletal muscle cells and stimulate the secretion of insulin after incubation of rat insulinoma (INS-1E beta) cells, even at concentrations as low as 10-12 to 10-8 M [17]. Such concentrations are likely to be achieved in subjects drinking unfiltered coffee, and may even be achieved from drinking filtered coffee.
Now the group has reported the results of the first studies to have been conducted into the antidiabetic effects of cafestol in a live animal model. They chose to study a group of 47 KKAy mice (a genetically modified strain that becomes obese shortly after birth and serves as a model for T2D). The mice were randomised to consume 1.1 mg (high), 0.4 mg (low) or zero (control) cafestol each day for 10 weeks.
The team found that in the groups consuming cafestol, fasting plasma glucose levels were 28-30% lower, and fasting levels of glucagon, the hormone that raises the concentration of glucose and fat in the bloodstream, were 20% lower than in the control group. When the researchers isolated pancreatic islets from the mice, they found that insulin secretion was increased by 75-87% in the cafestol group compared to the control group [18].
While their results suggest that cafestol can delay the development of T2D in KKAy mice, the team know that further studies will be required to determine whether or not cafestol can be of use in preventing or treating T2D in humans.
The background image is a Creative Commons licensed photograph of the fruits of yellow catuai coffee, a variety of Coffea arabica.
Reference(s)
- Clarke, R.J. and Macrae, R. (2013). Coffee Volume 1: Chemistry. Springer, New York. ISBN 978-9401086936.
- Yet more growth in UK coffee shop market as coffee shops become the new local UK Coffee Leader Summit News and Insight, 20 January 2017.
- Bøhn S.K., Blomhoff, R. and Paur, I. (2014) Coffee and cancer risk, epidemiological evidence, and molecular mechanisms. Mol. Nutr. Food Res., 58(5), 915–930.
- Grosso, G., Micek, A., Godos, J., Sciacca, S., Pajak, A., Martínez-González, M.A., Giovannucci, E.L. and Galvano, F. (2016) Coffee consumption and risk of all-cause, cardiovascular, and cancer mortality in smokers and non-smokers: a dose-response meta-analysis. Eur. J. Epidemiol., 31(12), 1191–1205.
- Slotta, K.H. and Neisser, K. (1938) Die Gewinnung von cafestolund anderen Verbindung aus dem Unverseifbaren des Kaffee-Öls.Chem. Ber., 71(9), 1991–1994.
- Slotta, K.H. and Neisser, K. (1938) Zur Konstitutionsaufklärung des Cafesterols. Chem. Ber., 71 (11), 2342–2346.
- Djerassi, C., Cais, M. and Mitscher, L.A. (1959) The structure of the pentacyclic diterpene cafestol. On the absolute configuration of diterpenes and alkaloids of the phyllocladene group. J. Am. Chem. Soc., 81(10), 2386–2398.
- Finnegan, R.A. and Djerassi, C. (1960) Further studies on the structure and absolute configuration of cafestol.J. Am. Chem. Soc., 82(16), 4342–4344.
- Cavin, C., Holzhäuser, D., Scharf, G., Constable, A., Huber, W.W. and Schilter, B. (2002) Cafestol and kahweol, two coffee specific diterpenes with anticarcinogenic activity. Food Chem. Toxicol., 40(8), 1155–1163.
- Cavin, C., Holzhäuser, D., Constable, A., Huggett, A.C. and Schilter, B. (1998) The coffee-specific diterpenes cafestol and kahweol protect against aflatoxin B1-induced genotoxicity through a dual mechanism. Carcinogenesis, 19(8), 1369–1375.
- Kim, J.Y., Jung, K.S. and Jeong, H.G. (2004) Suppressive effects of the kahweol and cafestol on cyclooxygenase-2 expression in macrophages. FEBS Lett., 569(1–3), 321–326.
- Lima, C.S., Spindola, D.G., Bechara, A., Garcia, D.M., Palmeira-Dos-Santos, C., Peixoto-da-Silva, J., Erustes, A.G., Michelin, L.F.G., Pereira, G.J.S., Smaili, S.S., Paredes-Gamero, E., Calgarotto, A.K., Oliveira, C.R. and Bincoletto, C. (2017). Cafestol, a diterpene molecule found in coffee, induces leukemia cell death. Biomed. Pharmacother., 92, 1045–1054.
- Thelle, D.S., Arnesen, E. and Førde, O.H. (1983) The Tromsø heart study. Does coffee raise serum cholesterol? N. Engl. J. Med., 308(24), 1454–1457.
- Heckers H, Göbel U, Kleppel U. (1994) End of the coffee mystery: diterpene alcohols raise serum low-density lipoprotein cholesterol and triglyceride levels. J. Intern. Med.,, 235(2), 192–193.
- Ding, M., Bhupathiraju, S.N., Chen, M., van Dam, R.M. and Hu, F.B. (2014) Caffeinated and decaffeinated coffee consumption and risk of type 2 diabetes: a systematic review and a dose-response meta-analysis. Diabetes Care, 37(2), 569–586.
- Zhang, C., Linforth, R. and Fisk, I.D. (2012) Cafestol extraction yield from different coffee brew mechanisms. Food Res. Int., 49(1), 27–31.
- Mellbye, F.B., Jeppesen, P.B., Hermansen, K. and Gregersen, S. (2015) Cafestol, a bioactive substance in coffee, stimulates insulin secretion and increases glucose uptake in muscle cells: studies in vitro. J. Nat. Prod., 37(2), 2447–2451.
- Mellbye, F.B., Jeppesen, P.B., Shokouh, P., Laustsen, C., Hermansen, K. and Gregersen, S. (2017) Cafestol, a bioactive substance in coffee, has antidiabetic properties in KKAy mice. J. Nat. Prod., 80(8), 2353–2359.
1st August 2017, (S)-Nicotine
Scientists have long known that birds inhabiting urban environments often use items of human litter to build their nests. In one instance, a species appears to have harnessed the toxic chemicals in discarded cigarette butts in an effort to ward off ectoparasites, such as ticks, from their nests. A team of researchers led by Constantino Macías Garcia at the National Autonomous University of Mexico noticed a peculiar behaviour in house finches (Carpodacus mexicanus) inhabiting the main campus of the university in Mexico City; they were lining their nests with toxic cigarette butts. The birds were actually tearing apart the paper that covers the cigarette filter and then separating the fibres. Initial studies suggested that (S)-nicotine (CHEBI:17688, the naturally occurring and active form of nicotine) and other chemicals present in the filters might help deter insect pests from moving into the nests, as nicotine is known to have antiparasitic properties, but the evidence was not conclusive [1].The team have recently published a more detailed follow-up study where they experimented with 32 house finch nests, wherein tick infestations could be controlled [2]. One day after the eggs in the nest had hatched, the researchers removed the natural nest lining and replaced it with artificial felt to ensure the removal of any parasites that might have moved in. They subsequently added live ticks to 10 of the nests, dead ticks to another 10 and left the remaining 12 nests free of ticks. They found that the adult finches were significantly more likely to add cigarette butt fibres to the nest if it contained ticks. In addition, the weight of cigarette butt material added to nests containing live ticks was 40% greater on average compared to that added to nests containing dead ticks. The results suggest that the finches are using the cigarette butts to self-medicate their nests against the ticks, which can cause damage to finches by eating their feathers and sucking their blood. While this behaviour has its benefits, the researchers have also discovered negative effects where the toxic chemicals present in the fibres cause genetic damage to the birds by interfering with cell division. Although the overall effect may be positive in the short term, researchers will need to conduct a long-term study to know exactly how living with cigarette butts ultimately impacts house finch population.
The background image is a detail from a Creative Commons licensed image depicting a male house finch.
Reference(s)
- Suárez-Rodríguez, M., López-Rull, I. and Macías Garcia, M. (2013). Incorporation of cigarette butts into nests reduces nest ectoparasite load in urban birds: new ingredients for an old recipe? Biol. Lett., 9, 20120931.
- Suárez-Rodríguez, M. and Macías Garcia, M. (2017). An experimental demonstration that house finches add cigarette butts in response to ectoparasites. J. Avian Biol., 48, doi:10.1111/jav.01324, published online 20 June 2017.
1st July 2017, (E)-1-hydroxy-2-(non-1-en-1-yl)quinolin-4-one
Most microorganisms occur not in colonies of single-species planktonic forms but in extremely complex polymicrobial communities in biofilms which may be attached to biotic or abiotic sites. For example, some 600 to 1,000 different species of bacteria are known to colonise the human mouth and the digestive tract [1,2]. As a consequence, many species-specific physical and chemical interactions have evolved between different species of microorganisms. These interactions may be mutualistic, synergistic, or antagonistic [3].The Gram-negative Pseudomonas aeruginosa and the Gram-positive Staphylococcus aureus can both cause a wide range of disease in humans, varying in severity from minor skin infections through to potentially fatal sepsis and endocarditis. Both species are often active in polymicrobial infections (that is, infections in which various combinations of viruses, bacteria, fungi, and parasites may be present) and interact in a competitive manner.
A well-studied example of this occurs in the lung disorder cystic fibrosis. S. aureus is usually the predominant pathogen colonising the lungs in the early stages of the disease. In later stages, P. aeruginosa prevails, although S. aureus is still present [4]. It is now known that P. aeruginosa makes use of two different approaches to contribute to the decline in S. aureus – one is by manipulation of the innate immunity of the host [5], while the other is to produce a number of antibacterial molecules known to inhibit competitors such as S. aureus [6].
The antibacterial compounds produced by P. aeruginosa include pyocyanine, cyanide, and a series of over 56 quinolones, 16 of which belong to the class of 2-alkyl-N-hydroxy-4-quinolones, in which the side-chains at position 2 are of varying length and have varying degrees of unsaturation [7]. Some of the compounds reported were produced in amounts of several milligrams per litre, while the concentration of others was orders of magnitude lower.
Now, Thomas Böttcher and Dávid Szamosvári at the University of Konstanz in Germany have reported total syntheses of the three most abundant 2-alkyl-N-hydroxy-4-quinolones. Two of these had saturated side chains (heptyl and nonyl); the third had a non-1-enyl chain in which the configuration (cis or trans) of the double bond was not known. Their synthetic strategy allowed them to control the stereochemistry of the side-chain double bond [8].
When tested against four different strains of S. aureus (including MRSA), Böttcher and Szamosvári found that the compound with the trans-nonenyl side chain, (E)-1-hydroxy-2-(non-1-en-1-yl)quinolin-4-one (CHEBI:137441), was up to 20-times more active than the compounds with saturated side chains. In contrast, they found that the cis-isomer was inactive.
The researchers suggest that (E)-1-hydroxy-2-(non-1-en-1-yl)quinolin-4-one is likely to be a major factor in cross-species interactions during polymicrobial infections and that the significant differences in the activity between the three major 2-alkyl-N-hydroxy-4-quinolones produced by P. aeruginosa may imply that the less active compounds are playing other, as yet unknown roles that contribute to the microorganism's survival.
The background image is a Creative Commons licensed photograph of Pseudomonas aeruginosa, Enterococcus faecalis and Staphylococcus aureus on tryptic soy agar.
Reference(s)
- Aas, J.A., Paster, B.J., Stokes, L.N., Olsen, I. and Dewhirst, F.E. (2005) Defining the normal bacterial flora of the oral cavity. J. Clin. Microbiol., 43(11), 5721–5732.
- Manson, J.M., Rauch, M. and Gilmore, M.S. (2008) The commensal microbiology of the gastrointestinal tract. Adv. Exp. Med. Biol., 635, 15–28.
- Peters, B.M., Jabra-Rizk, M.A., O'May, G.A., Costerton, J.W. and Shirtliff, M.E. (2012) Polymicrobial interactions: impact on pathogenesis and human disease. Clin. Microb. Rev., 25(1), 193–293.
- Baldan, R., Cigana, C., Testa, F., Bianconi, I., De Simone, M., Pellin, D., Di Serio, C., Bragonzi, A. and Cirillo, D.M. (2014) Adaptation of Pseudomonas aeruginosa in cystic fibrosis airways influences virulence of Staphylococcus aureus in vitro and murine models of co-infection. PLoS One, 9(3), e89614.
- Pernet, E., Guillemot, L., Burgel, P.R., Martin, C, Lambeau, G., Sermet-Gaudelus, I., Sands, D., Leduc, D., Morand, P.C., Jeammet, L., Chignard, M., Wu, Y. and Touqui, L. (2014) Pseudomonas aeruginosa eradicates Staphylococcus aureus by manipulating the host immunity. Nat. Commun., 5, 5105.
- Filkins, L.M., Graber, J.A., Olson, D.G., Dolben, E.L., Lynd, L.R., Bhuju, S. and O'Toole, G.A. (2015) Coculture of Staphylococcus aureus with Pseudomonas aeruginosa drives S. aureus towards fermentative metabolism and reduced viability in a cystic fibrosis model. J. Bacteriol., 197(14), 2252–2264.
- Lépine, F., Milot, S., Déziel, E., He, J. and Rahme L,.G. (2014) Electrospray/mass spectrometric identification and analysis of 4-hydroxy-2-alkylquinolines (HAQs) produced by Pseudomonas aeruginosa. J. Am. Soc. Mass Spectrom., 55(6), 862–869.
- Szamosvári, D. and Böttcher, T. (2017) An unsaturated quinolone N-oxide of Pseudomonas aeruginosa modulates growth and virulence of Staphylococcus aureus. Angew. Chem. Int. Ed. Engl., 56(25), 7271–7275.
1st June 2017, JQ1
Heart failure continues to be a common and difficult-to-treat medical condition with a high mortality rate. Despite widespread use of approved drugs, approximately 40% of patients with heart failure die within five years of their initial diagnosis, hence there is a great need for more effective therapeutics. Existing heart failure drugs consist of β-adrenergic receptor antagonists (commonly known as β-blockers) and acetylcholine esterase inhibitors, which act by blocking stress hormones at the surface of heart cells.Now a team of researchers led by Saptarsi Haldar MD, at the Gladstone Institutes in San Francisco have discovered that newly-developed cancer drugs may also provide an alternative treatment for heart failure patients [1]. In previous studies, the team observed that an anti-cancer drug candidate called JQ1 (CHEBI:137113) can also prevent the development of heart failure in mouse models when administered at the very onset of the disease [2]. However, as the majority of patients requiring treatment already have longstanding cardiac dysfunction, the team decided to investigate whether JQ1 could also be used to treat established heart failure.
JQ1 is part of a class of drugs that act by inhibiting a protein called bromodomain-containing protein 4 (BRD4), which is a common factor in both the development of tumours and the onset of inflammation and fibrosis (scarring) in heart cells, two of the main causes of heart failure. Initial studies using mouse models showed that JQ1 improved all the hallmark features of heart failure. To determine whether JQ1 would have the same effect in humans, the drug was tested on cardiomyocytes, a type of beating cell generated from adult human skin cells. Results were encouraging and consistent with the mouse model studies. In contrast to several cancer drugs that cause cardiotoxicity, these studies suggest that BRD4 inhibitors may be a special class of anti-cancer agents that has cardioprotective properties. Given that drug candidates derived from JQ1 are currently being tested in cancer clinical trials, their safety in humans is already being determined. This key information could accelerate the development of a new heart failure drug and make it available to patients more quickly.
The background image is a detail from a Creative Commons licensed image depicting a human heart diagram from the textbook 'Outlines of Human Physiology; Designed for the Use of the Higher Classes in Common Schools' published in 1834.
Reference(s)
- Duan, Q., McMahon, S., Anand, P., Shah, H., Thomas, S., Salunga, H.T., Huang, Y., Zhang, R., Sahadevan, A., Lemieux, M.E., Brown, J.D., Srivastava, D., Bradner, J.E., McKinsey, T.A. and Haldar, S.M. (2017). BET bromodomain inhibition suppresses innate inflammatory and profibrotic transcriptional networks in heart failure. Sci. Transl. Med., 9, doi:10.1126/scitranslmed.aah5084, published online 17 May 2017.
- Anand, P., Brown, J.D., Lin, C.Y., Qi, J., Zhang, R., Artero, P.C., Alaiti, M.A., Bullard, J., Alazem, K., Margulies, K.B., Cappola, T.P., Lemieux, M., Plutzky, J., Bradner, J.E. and Haldar, S.M. (2013). BET bromodomains mediate transcriptional pause release in heart failure. Cell, 154,569–582.

1st May 2017, Bisphenol A
Bisphenol A (also known as BPA, CHEBI:33216) was first synthesised in 1891 [1], but it received little attention for over forty years. In the mid-1930s, Edward Charles Dodds, Professor of Biochemistry at the University of London, reported BPA among the first group of synthetic xenoestrogens that did not have a phenanthrene skeleton [2].Later in the 1950s, BPA found use in the production of a hard plastic known as polycarbonate, which was strong enough to replace steel and clear enough to replace glass. Finding use in applications as diverse as food containers, electronics, safety equipment, and motor vehicles, the production and use of BPA soared, with several million tonnes being manufactured each year [5].
As BPA production increased, attention turned to its possible effects on the large number of workers who were potentially exposed to it. During the 1970s and 1980s, several studies led to the conclusion that there was no convincing evidence of its carcinogenicity. In the 1990s, however, the estrogenic properties of BPA came in for further investigation [6].
In 1993, a team of endocrinologists at Stanford University who were searching for an endogenous estrogen in yeast realised that the estrogenic substance they had isolated was actually BPA that had leached from polycarbonate flasks in their lab [7]. Their findings brought BPA to the attention of researchers in the area of endocrine disruption – the idea that some chemicals interfere with the production and transmission of hormones in the body and so disrupt the normal functioning of the endocrine system – fuelling studies into the possibility that BPA could leach into the environment and act as a xenoestrogen.
More than twenty years on, the evidence that BPA is a xenoestrogen at exposure levels at or below the existing safety standards remains inconclusive. However, pressure from consumer groups and retailers has caused the plastics industry to search for alternatives for BPA, particularly in products that come into contact with food and beverages. One such substitute is 9,9-bis(4-hydroxyphenyl)fluorene (BHPF), which in recent years has been used as a replacement for BPA in the production of numerous products, including protective coatings, moulded products, and structural adhesives.
Now, a study by a team of Chinese and Japanese scientists suggests that BHPF may not appropriate as a replacement for BPA in the production of plastics for food containers or water bottles, however [8]. The researchers found that not only is BHPF released from commercial 'BPA-free' plastic bottles into drinking water (they found BHPF in the plasma of seven out one hundred college students who habitually used plastic water bottles), but that is a powerful anti-estrogen that can cause adverse pregnancy outcomes in mice. The researchers recommend that as well as being checked for estrogenic activity, possible substitutes for BPA should be also be checked for anti-estrogenic activity.
The background image is a Creative Commons licensed photograph of a polycarbonate water bottle.
Reference(s)
- Dianin, A.P. (1891) On condensation products of ketones with phenols. Zh. Russ. Fiz.-Khim. O-va.,23(2), 488–517, 523–546, 601–611.
- Dodds, E.C. and Lawson, W, (1936) Synthetic Œstrogenic agents without the phenanthrene nucleus. Nature, 137(3476), 996.
- Dodds later found that diethylstilbestrol (DES) was a vastly more powerful estrogen and for over three decades, DES was used to treat problems relating to menstruation, morning sickness, and the prevention of miscarriages; it was also used in farming to increase meat production. As a result of concerns about its carcinogenicity, DES was eventually banned from use in humans and in meat production during the 1970s.
- American Chemistry Council Information Sheet About BPA: Epoxy Resins, October 2016.
- World BPA production grew by over 372,000 tonnes in 2012. Merchant Research & Consulting Ltd. News release, November 8 2013.
- Vogel, S.A. (2009) The politics of plastics: the making and unmaking of bisphenol A "safety". Am. J. Public Health, 99(Suppl. 3), S559–S566.
- Krishnan, A.V., Stathis, P., Permuth, S.F., Tokes, L. and Feldman, D. (1993) Bisphenol-A: an estrogenic substance is released from polycarbonate flasks during autoclaving. Endocrinology, 132(6), 2279–2286.
- Zhang, Z., Hu, Y., Guo, J., Yu, T., Sun, L., Xiao, X., Zhu, D., Nakanishi, T., Hiromori, Y., Li, J., Fan, X., Wan, Y., Cheng, S., Li, J., Guo, X. and Hu J. (2012) Fluorene-9-bisphenol is anti-oestrogenic and may cause adverse pregnancy outcomes in mice. Nat. Commun., 8(11), 14585.
1st April 2017, Pentamidine
In February 2017, the World Health Organisation (WHO) published a list of the 12 most dangerous bacterial pathogens for which new antibiotics are urgently required [1]. It is dominated by Gram-negative bacteria, which have become resistant to multiple antibiotics and represent a significant danger to public health.Gram-negative bacteria have the common feature of two sets of outer membranes that form a shell to protect their innards and act as a barrier to many otherwise effective antibiotics. The bacteria can cause life-threatening pneumonia or systemic infections, and patients are increasingly becoming infected in hospitals. As a last resort, the infections can be treated with the antibiotic colistin, which is toxic to both kidney and nerve cells, but some bacteria have developed resistance even to this.
To tackle this problem, a team of researchers led by Professor Eric Brown at McMaster University in Canada have investigated molecules that might weaken the outer membranes so that previously ineffective antibiotics can penetrate the cells and kill the bacteria. The team screened a collection of 1,440 previously approved drugs against a strain of E. coli and found the antifungal and trypanocidal drug pentamidine (CHEBI:45081) to be particularly effective in penetrating the outer membranes [2]. In vitro studies indicated that when combined with pentamidine, antibiotics typically restricted to Gram-positive bacteria, such as rifampicin and novobiocin, were effective against a range of Gram-negative bacteria. Further experiments were conducted with mice infected with colistin-resistant Acinetobacter baumannii, a pathogen rated by the WHO as a 'critical priority' on their list of dangerous bacteria. They found that 10 out of 11 mice showed no signs of infection 7 days after treatment with a combination of pentamidine and novobiocin. Although pentamidine has some toxicity issues, the doses required in mice were relatively low. The new work supports a growing belief among scientists that developing compounds to weaken rather than kill bacteria can lessen pathogens' evolutionary drive to become resistant. The research team are continuing to use their screening platform to search for other molecules with outer-membrane weakening properties.
The background image is a detail from a Creative Commons licensed image depicting a colourised scanning electron micrograph of E. coli.
Reference(s)
- Global Priority List of Antibiotic-Resistant Bacteria to Guide Research, Discovery, and Development of New Antibiotics. (World Health Organisation, Geneva, 27 February 2017
- Stokes, J.M., MacNair, C.R., Ilyas, B., French, S., Côté:, J.P., Bouwman, C., Farha, M.A., Sieron, A.O., Whitfield, C., Coombes, B.K. and Brown, E.D. (2017). Pentamidine sensitizes Gram-negative pathogens to antibiotics and overcomes acquired colistin resistance. , doi:Nat. Microbiol., 2, 17028 (published online 06 March 2017).
1st March 2017, Hypoglycin A
For at least twenty years in Bihar, the third most populous state in India, there have been reports of previously healthy children waking in the night during the early summer convulsed with seizures and suffering from mental confusion and memory loss. In around a third of cases, they then fall into a coma and die; many of the survivors are left brain damaged. Each year, the outbreak of cases ends suddenly with the arrival of the monsoon rains in July, only to return again the following summer.Doctors investigated the possibility of various viruses as well as infections by mosquitoes, rats or bats but were unable to identify the cause. They came closer to the solution than they realised when they looked into possible effects of pesticides in the lychee (also known as litchi, Litchi chinensis) orchards that are widespread in Bihar, but were still unable to explain the outbreaks. The Indian authorities believed that the illness was probably a type of viral encephalitis. In 2013 and 2014, nearly 2 million children in Bihar were vaccinated against Japanese encephalitis virus, which is transmitted by mosquitoes.
The puzzle was eventually solved after a team from the National Centre for Disease Control (NCDC) in Delhi, led by Aakash Shrivastava, initiated a joint investigation with a team led by Padmini Srikantiah from the US Centers for Disease Control and Prevention (US CDC). In 2013, they studied over 130 children admitted to to two hospitals in Muzaffarpur.
The investigators found no evidence of viruses or brain inflammation. An early finding was that on admission to hospital, many victims had very low blood sugar levels, which was also associated with increased mortality. In June 2013, NCDC advised Muzaffarpur health officials to raise the blood sugar levels of patients by administering intravenous dextrose.
The following year the teams focussed on another set of children aged 15 or under who were admitted between 26th May and 17th July. Of the 390 cases, 122 died, making the mortality rate in 2014 almost a third lower than the previous year. This suggested the possibility that the victims had been exposed to an environmental toxin that caused the low blood glucose levels and resulted in the observed seizures and encephalopathy.
Lab investigations found that none of the sick children – even those who died from the illness – had a high white blood cell count, implying that they were not fighting an infection. The teams also noticed that, unlike a normal epidemic, the disease did not strike in clusters. "Each affected child seemed to be an isolated case in a village," the researchers reported.
Finding no evidence of a known infectious cause, the teams examined various possible non-infectious causes, including pesticides or herbicides used to spray orchards and fields, insecticides used in vector-borne disease control measures, heavy metals, and exposure to unusual medications. All proved negative.
Searching in PubMed, the researchers found previous reports of a vomiting sickness that had struck Jamaicans for decades. The Jamaica outbreaks had eventually been shown to be caused by the toxins hypoglycin A (CHEBI:134622) and hypoglycin B found in unripe ackee (Blighia sapida) fruit, and were stopped after public health warnings not to eat the unripe fruit [1]. The hypoglycins are so-named because of their hypoglycaemic activity – they inhibit fatty acid metabolism, which in turn blocks glucose production in cells.
The ackee is a member of the same botanical family as the lychee, which is widely grown in Bihar – around 70% of India's lychee crop comes from Bihar. Unripe lychees also contain hypoglycin A, as well as a closely related toxin, L-α-(methylenecyclopropyl)-glycine (MCPG) [2]. Was it possible that unripe lychees could be responsible for the Bihar outbreak?
The teams detected the presence of metabolites of hypoglycin A and/or MCPG in the urine of 66% of the illness victims, but none in an age-matched control set. They also found that 90% of the patients had abnormal plasma acylcarnitine profiles – consistent with severe disruption of fatty acid metabolism.
While the investigators could demonstrate an association between the outbreak illness, lychee consumption, and both hypoglycin A and MCPG toxicity, they were unable to prove definitively that the former was caused by the latter. However, assessing their results using the Bradford Hill criteria for causation showed that seven of the nine criteria were met. In a recently published account of their work [3], they conclude that their findings "reflect a plausible, but not necessarily sufficient, causal pathway between litchi consumption and illness".
To prevent the illness and reduce mortality the teams recommend minimising the consumption of lychees and ensuring that children have an evening meal throughout the outbreak period, since missing the evening meal is more likely to result in night-time hypoglycaemia, particularly in young children. Where cases are suspected, the teams recommend checking blood glucose levels and, where low levels are found, giving corrective amounts as necessary.
There are still some unanswered questions. For example, while young children often spend their day eating lychees in the orchards that surround many villages in Bihar, why is (typically) only one child in an entire village struck by the illness? Many of those struck down by the illness were malnourished (Bihar is one of the poorest states in India). It may be that such a child with low glycogen and glucose stores is particularly vulnerable to lychee toxins. The teams suggest that genetic factors might also be involved.
The lychee toxin mechanism would appear to explain similar illnesses reported in lychee-growing regions of Bangladesh and northern Vietnam [4], although there are differences. In Vietnam, for example, where the disease is known as "Ac Mong encephalitis", after the Vietnamese term for nightmare, the symptoms start with headache and fever, which is not the case for the victims in India. Perhaps levels of different toxins vary in lychees (as they do in ackee fruit) depending on factors such as the cultivar that is being grown, the soil conditions, and the ripeness of the fruit.
The background image is a Creative Commons licensed photograph of a botanical plate of a lychee tree and fruit, together with its name in Latin and Chinese, in Flora Sinensis (1656), one the first European works on the natural history of China. It was published in Vienna by the Jesuit missionary and scientist Father Michael Piotr Boym (1612–1659), who spent over a decade in China.
Flora Sinensis is believed to be the first book to use the name "Flora" to mean the plants of particular area. However, as well as seventeen botanical plates of cultivated fruits of south-eastern China, together with a Latin description of each plant and its medicinal properties, it also includes five plates of animals (including one of a hippopotamus) and one plate depicting a Chinese stele. Originally published uncoloured, hand-coloured copies of Flora Sinensis are incredibly rare; one is held by the library of the Natural History Museum in London.
Reference(s)
- Hassall, C.H. and Reyle, K. (1955), Hypoglycin A and B, two biologically active polypeptides from Blighia sapida. Biochem J. 1955, 60(2), 334–339.
- Gray, D.O. and Fowden, L., (1962) α-(Methylenecyclopropyl)glycine from Litchi seeds. Biochem J. 1962, 82, 385–389.
- Shrivastava, A., Kumar, A., Thomas, J.D., Laserson, K.F., Bhushan, G., Carter, M.D., Chhabra, M., Mittal, V., Khare, S., Sejvar, J.J., Dwivedi, M., Isenberg, S.L., Johnson, R., Pirkle, J.L., Sharer, J.D., Hall, P.L., Yadav, R., Velayudhan, A., Papanna, M., Singh, P., Somashekar, D., Pradhan, A., Goel, K., Pandey, R., Kumar, M., Kumar, S.,Chakrabarti, A., Sivaperumal, P., Kumar, A.R., Schier, J.G., Chang, A., Graham, L.A., Mathews, T.P., Johnson, D., Valentin, L., Caldwell, K.L., Jarrett, J.M., Harden, L.A., Takeoka, G.R., Tong, S., Queen, K., Paden, C., Whitney, A., Haberling, D.L., Singh R., Singh R.S., Earhart K.C., Dhariwal A.C., Chauhan, L.S., Venkatesh, S. and Srikantiah, P. (2017) Association of acute toxic encephalopathy with litchi consumption in an outbreak in Muzaffarpur, India, 2014: a case-control study. Lancet Global Health, DOI: 10.1016/S2214-109X(17)30035-9 (In Press, corrected proof published online 31 Jan 2017).
- Paireau, J., Tuan, N.H., Lefrançois, R., Buckwalter, M.R., Nghia, N.D., Hien, N.T., Lortholary, O., Poirée, S., Manuguerra, J.-C., Gessain, A., Albert, M.L., Brey, P.T., Nga, P.T. and Fontanet, A. (2012) Litchi–associated acute encephalitis in children, Northern Vietnam, 2004–2009. Emerging Infect. Dis., 18(11), 1817–1824.
1st February 2017, Luteolin
Breast cancer is the most common cancer in women and is known to cause around half a million deaths worldwide per year. Triple-negative breast cancers (TNBC), which comprise 15-20% of all breast tumours, are particularly deadly and are so named because they fail to express the three key receptors targeted by current chemotherapy regimens [1]. As a result, common cancer drugs are unable to locate the cells and hence death usually occurs following metastasis of TNBC cells and development of tumours at secondary sites.Generally, TNBC can only be treated using extremely aggressive and highly toxic non-targeted drug combinations, hence the search is on for safer and more effective treatments. Recent studies conducted by a team led by Professor Salman Hyder at the University of Missouri have focused on luteolin (CHEBI:15864), a naturally occurring, non-toxic compound found in herbs such as thyme and parsley, and vegetables such as celery and broccoli, could reduce the risk of developing metastasis originating from TNBC in women. Numerous studies have reported that luteolin induces apoptosis (programmed cell death) in various types of cancer cells, including breast cancer [2]. Using human TNBC cells grown in mice, the research team tested luteolin to determine if it could suppress metastasis. In the first series of tests, the team observed that luteolin inhibited the metastasis of TNBC in the lungs of affected mice with no obvious toxic effects [3]. In further tests the scientists found that pre-teatment of in vitro-cultured TNBC was found to inhibit cell migration in a dose-dependent manner. Although early-stage results of this research are promising, additional studies to determine the effect of the compound in humans are required.
The background image is a detail from a Creative Commons licensed image depicting part of a broccoli flowerhead, which is a rich source of dietary flavonoids including luteolin.
Reference(s)
- Lehmann, B.D., Bauer, J.A., Chen, X., Sanders, M.E., Chakravarthy, A.B., Shyr, Y. and Pietenpol, J.A. (2011). Identification of human triplenegative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Invest., 121, 2750–2767..
- Cook, M.T., Liang, Y., Besch-Williford, C., Goyette, S., Mafuvadze, B. and Hy1der, S.M. (2015). Luteolin inhibitsprogestin-dependent angiogenesis, stem cell-like characteristics, and growth of human breast cancer xenografts. SpringerPlus, 4, 444–459.
- Cook, M.T., Liang, Y., Besch-Williford, C. and Hy1der, S.M. (2017). Luteolin inhibits lung metastasis, cell migration, and viability of triple-negative breast cancer cells. Breast Cancer: Targets Ther., 9, 9–19.

1st January 2017, S,S-dimethyl-beta-propiothetin
Millions of tons of plastic waste are dumped into the sea every year and plastic debris is found in pelagic environments worldwide [1]. However, a 2014 analysis estimated that there was only around 250,000 tonnes of plastics suspended in the world's oceans [2,3]. The fate of the 'missing' plastic is still a topic of study and debate. Whatever the eventual findings, the consequences of plastics pollution in the oceans on marine life are grim.Although the Midway Atoll is one of the world's most remote marine sanctuaries, nearly all of the 1.5 million Laysan albatrosses that nest there have plastic in their digestive system. Around a third of the chicks born there die as a result of being fed lethal quantities of plastic by their parents, who mistake the floating debris for food as they forage over the polluted Pacific Ocean [4].
The albatrosses are not alone in mistaking the plastic for food. Hundreds of species of marine fish, marine mammals, sea turtles and seabirds ingest plastic debris, but quite why is often unknown. While it is generally assumed that they visually mistake the plastic debris for prey, this is not necessarily the case. Now a team led by Matthew Savoca, a graduate student working with professor Gabrielle Nevitt at the University of California, Davis, has shown that, for the albatrosses at least, the reason is not appearance but smell – the plastics smell like food.
The team took new beads made of the three common types of plastic debris: high-density polyethylene (HDPE), low-density polyethylene (LDPE), and polypropylene (PP). Mesh bags containing the beads were attached to two buoys off the Californian coast. Three weeks later, the samples were retrieved and the headspace sulfur volatiles were isolated by solid-phase microextraction and identified by gas chromatography using a sulfur chemiluminescence detector.
They found that gas from the headspace around all of the sea-soaked samples contained dimethyl sulfide (DMS, CHEBI:17437), whereas that from the headspace around untreated plastic beads did not [5]. The findings show that within a few days of entering the water, the plastic debris becomes covered in marine phytoplankton. During grazing by krill, the phytoplankton produce DMS by enzymatic breakdown of dimethylsulfoniopropionate (DMSP, CHEBI:16457) [6].
Albatrosses are examples of procellariiformes, also known as tubenoses, an order of seabirds whose members share a keen sense of smell. DMS indicates to the albatrosses that krill – the birds' prey – are nearby and triggers foraging activity in the albatrosses and in other marine organisms [7-9]. The concentrations of DMS measured around the plastic beads by Savoca's group were several orders of magnitude greater than those found where DMS-responsive procellariiform species have been reported to aggregate.
The team's findings suggest that by emitting the scent of a marine infochemical produced by the DMSP/DMS pathway, the plastic debris becomes an olfactory trap for susceptible marine wildlife. The team believe their work may also help to explain plastic ingestion patterns found in other groups, including sea turtles, penguins, various species of fish, and marine mammals, all of which either detect or use these compounds during foraging.
The background image is a Creative Commons licensed photograph of a Laysan Albatross (Phoebastria immutabilis) taken at Kilauea Lighthouse on Kauai, Hawaii.
Reference(s)
- Jambeck, J.R., Geyer, R., Wilcox, C., Siegler, T.R., Perryman, M., Andrady, A., Narayan, R. and Law, K.L. (2015) Marine pollution. Plastic waste inputs from land into the ocean. Science, 347(6223), 768–771.
- Cózar, A., Echevarría, F., González-Gordillo, J.I., Irigoien, X., Úbeda, B., Hernández-León, S. Palma, A.T., Navarro, S., García-de-Lomas, J. Ruiz, A. Fernández-de-Puelles, M.L. and Duarte, C.M. (2014) Plastic debris in the open ocean. Proc. Natl. Acad. Sci. U. S. A., 111(28), 10239–10244.
- Eriksen, M., Lebreton, L.C.M., Carson, H.S., Thiel, M., Moore, C.J., Borerro, J.C., Galgani, F., Ryan, P.G. and Reisser, J. (2014) Plastic pollution in the world's oceans: more than 5 trillion plastic pieces weighing over 250,000 tons afloat at sea.PLoS One, 9, e111913.
- Jordan, C. (2009) Midway: Message from the Gyre N. Y. Rev. Books, Nov. 11.
- Savoca, M.S., Wohlfeil, M.E., Ebeler, S.E. and Nevitt, G.A. (2016) Marine plastic debris emits a keystone infochemical for olfactory foraging seabirds. Sci. Adv., 2(11), e1600395.
- Dacey, J.W. and Wakeham, S.G. (1986) Oceanic dimethylsulfide: production during zooplankton grazing on phytoplankton. Science (Washington, DC, U. S.), 233(4770), 1314–1316.
- Nevitt, G.A., Veit, R.R. and Kareiva, P. (2002) Dimethyl sulphide as a foraging cue for Antarctic Procellariiform seabirds. Nature, 376, 680–682.
- Savoca M.S. and Nevitt G.A. (2014) Evidence that dimethyl sulfide facilitates a tritrophic mutualism between marine primary producers and top predators. Proc. Natl. Acad. Sci. U. S. A., 111(11), 4157–4161.
- Entity of the Month 7th April 2014, Dimethyl sulfide.
2016

1st December 2016, Aspartame
Aspartame (CHEBI:2877) is a common sugar substitute used as a sweetener in many prepared foods and beverages, particularly diet drinks. It is a common choice for those trying to lose weight, as it contains reduced calories and decreases the incidence of metabolic syndrome (the generic term for a group of symptoms associated with type 2 diabetes and cardiovascular disease). However, sometimes people who consume diet soft drinks actually end up putting on more weight and developing diabetes. This has continued to puzzle nutritionists, but recent research by a team led by Richard Hodin at Massachusetts General Hospital has found a possible mechanism to explain why aspartame is not always effective in promoting weight loss. They found that aspartame interferes with the action of an enzyme present on the human gut called intestinal alkaline phosphatase (IAP). This enzyme is primarily produced in the small intestine and has been shown to have protective effects towards metabolic syndrome [1].In an earlier study, the team had fed IAP to mice on a high fat diet and found that this not only prevented the onset of metabolic syndrome but also reduced the symptoms in mice that already had the condition [2]. One of the major metabolites of aspartame is phenylalanine, a known inhibitor of IAP [3]. The team proposed that that aspartame consumption might contribute to the development of metabolic syndrome due to inhibition of endogenous IAP by phenylalanine. In their current study, the team first found that the activity of IAP was reduced when the enzyme was added to a solution containing an aspartame-sweetened soft drink but remained unchanged if added to a solution with a sugar-sweetened beverage. When the team subsequently injected aspartame into segments of mouse intestine, levels of IAP fell sharply by around 50% [4]. The researchers followed four groups of mice for 18 weeks. Two groups were fed a normal diet, one receiving drinking water laced with aspartame, the other receiving plain water. The other two groups were fed a high-fat diet, together with either aspartame solution or plain water. The amount of aspartame administered to the mice was the equivalent to an adult human drinking approximately three cans of diet beverage on a daily basis. At the end of the study period, while there was negligible weight difference between the two groups fed a normal diet, mice on a high-fat diet/aspartame combination gained significantly more weight than those on the same diet that received plain water. In addition the aspartame-receiving mice in both diet groups displayed higher blood sugar levels compared with the other two groups, an early sign of the onset of diabetes. It remains to be seen whether what has been observed in mice will also apply to humans.
The background image is a detail from a Creative Commons licensed image depicting a batch of homemade sugar-free chocolate.
Reference(s)
- Economopoulos, K.P., Ward, N.L., Phillips, C.D., Teshager, A., Patel, P., Mohamed, M.M., Hakimian, S., Cox, S.B., Ahmed, R., Moaven, O., Kaliannan, K., Alam, S.N., Haller, J.F., Goldstein, A.M., Bhan, A.K., Malo, M.S. and Hodin, R.A. (2016). Prevention of antibiotic-associated metabolic syndrome in mice by intestinal alkaline phosphatase. Diabetes, Obes. Metab., 18, 519–527.
- Kaliannan, K., Hamarneh, S.R., Economopoulos, K.P., Nasrin Alam, S., Moaven, O., Patel, P., Malo, N.S., Ray, M., Abtahi, S.M., Muhammad, N., Raychowdhury, A., Teshager, A., Mohamed, M.M., Moss, A.K., Ahmed, R,, Hakimian, S., Narisawa, S., Millán, J.L., Hohmann, E., Warren, H.S., Bhan, A.K., Malo, M.S. and Hodin, R.A. (2013). Intestinal alkaline phosphatase prevents metabolic syndrome in mice. Proc. Natl. Acad. Sci. U. S. A., 110, 7003–7008.
- Fishman, W.H., Green, S. and Inglis, N.I. (1963). L-Phenylalanine: an organ specific, stereo-specific inhibitor of human intestinal alkaline phosphatase. Nature, 198, 685–686.
- Gul, S.S., Hamilton, A.R.L., Munoz A.R., Phupitakphol, T., Liu, W., Hyoju S.K., Economopoulos, K.P., Morrison, S., Hu, D., Zhang, W., Gharedaghi, M.H., Huo, H., Hamarneh, S.R., and Hodin, R.A. (2016). Inhibition of the gut enzyme intestinal alkaline phosphatase may explain how aspartame promotes glucose intolerance and obesity in mice. Appl. Physiol., Nutr., Metab., doi:10.1139/apnm-2016-0346, published online 18 November 2016.
1st November 2016, GNF6702
Chagas disease, leishmaniasis and African trypanosomiasis (sleeping sickness) affect 20 million people worldwide, primarily in Africa, Asia and South America and are known to cause more than 50,000 deaths annually [1]. The diseases are caused by infection, via insect bites, with the kinetoplastid parasites Trypanosoma cruzi, Leishmania spp. and Trypanosoma brucei spp. respectively. Current treatments for the three diseases are expensive, have adverse side effects and are not always very effective. In some cases, side-effects are so acutely toxic that up to a fifth of people cease treatment before therapy is complete. The kinetoplastid parasites that cause these diseases are a type of single-celled organism and are known to share similar biology and genetics [2]. This has led scientists to investigate the possibility of a single treatment for all three diseasesIn a recent study funded by the Wellcome Trust, a team of researchers led by Frantisek Supek at the Genomics Institute of the Novartis Research Foundation in San Diego, California has identified a potential solution to this problem [3]. The team screened around 3 million compounds in proliferation assays and identified GNF5343 to be effective against the parasites but harmless to mammalian cells. After further structural modifications of GNF5343 the team identified a lead compound, GNF6702 (CHEBI:133824), which displayed a 400-fold increase in potency compared to GNF5343. In tests with mice infected with the three parasites, GNF7602 showed a level of potency comparable with that of existing medicines but with minimal side-effects. The drug works by specifically inhibiting the kinetoplastid proteasome through a non-competitive mechanism, while being completely harmless to mammalian proteasomes. GNF7602 is currently undergoing toxicology evaluation before it can be advanced to trials in humans.
The background image is a Creative Commons licensed picture of Triatoma brasiliensis, which acts as a vector for Chagas disease.
Reference(s)
- Research priorities for Chagas disease, human African trypanosomiasis and leishmaniasis (2012). World Health Organization, WHO Technical Report Series, 975, 1–100.
- El-Sayed, N.M., Myler, P.J., Blandin, G., Berriman, M., Crabtree, J., Aggarwal, G., Caler, E., Renauld, H., Worthey, E.A., Hertz-Fowler, C., Ghedin, E., Peacock, C., Bartholomeu, D.C., Haas, B.J., Tran, A.N., Wortman, J.R., Alsmark, U.C., Angiuoli, S., Anupama, A., Badger, J., Bringaud, F., Cadag, E., Carlton, J.M., Cerqueira, G.C., Creasy, T., Delcher, A.L., Djikeng, A., Embley, T.M., Hauser, C., Ivens, A.C., Kummerfeld, S.K., Pereira-Leal, J.B., Nilsson, D., Peterson, J., Salzberg, S.L., Shallom, J., Silva, J.C., Sundaram, J., Westenberger, S., White, O., Melville, S.E., Donelson, J.E., Andersson, B., Stuart, K.D. and Hall, N. (2005). Comparative genomics of trypanosomatid parasitic protozoa. Science, 309, 404–409.
- Khare, S., Nagle, A.S., Biggart, A., Lai, Y.H., Liang, F., Davis, L.C., Barnes, S.W., Mathison, C.J., Myburgh, E., Gao, M.Y., Gillespie, J.R., Liu, X., Tan, J.L., Stinson, M., Rivera, I.C., Ballard, J., Yeh, V., Groessl, T., Federe, G., Koh. H.X., Venable, J.D., Bursulaya, B., Shapiro, M., Mishra, P.K., Spraggon, G., Brock, A., Mottram, J.C., Buckner, F.S., Rao, S.P., Wen, B.G., Walker, J.R., Tuntland, T., Molteni, V., Glynne, R.J. and Supek, F. (2016). Proteasome inhibition for treatment of leishmaniasis, Chagas disease and sleeping sickness. Nature 537, 229–233.
1st October 2016, Taurine
The amino sulfonic acid taurine (CHEBI:15891, also known as 2-aminoethanesulfonic acid) gets its name from taurus, the Latin name for bull or ox, as it was from ox bile that the compound was first isolated by Friedrich Tiedemann (1781–1861) and Leopold Gmelin (1788–1853) while working at the University of Heidelberg in 1827 [1]. Among the general public it is probably best known as an additive in many energy drinks, but while some studies suggest that taurine supplements may improve athletic performance, a 2008 review concluded that the amount of taurine present in these drinks was "far below the amounts expected to deliver either therapeutic benefits or adverse events" [2]. Taurine is also known as an additive in some contact lens solutions – it inhibits protein denaturation, so may decrease protein binding to contact lenses [3,4].Actually, taurine is vastly more important than most people realise. Synthesised in the body from methionine or cysteine, it is widely distributed throughout animal tissues – it makes up around 0.1% of human body weight – and is able to cross the blood-brain barrier. Taurine has many biological properties, acting as an antioxidant, an osmoregulation agent and as a substrate for formation of bile acid conjugates. It is essential for cardiovascular function as well as the development and function of the central nervous system, skeletal muscle, and the retina. An abundant component of meat and fish-based foods, it has been used as an oral supplement in the treatment of disorders as diverse as cystic fibrosis [5] and hypertension [6].
Recently, the importance of taurine in the supply of fish has been making news. According to the latest report by the Food and Agriculture Organisation of the United Nations, while global per capita fish consumption has risen above 20 kg per year for the first time, nearly a third of commercial fish stocks are now fished at biologically unsustainable levels – three times the level of 1974 [7]. In Japan, where fish consumption is closer to 70 kg per person per year, there has been an understandable increase in the use of aquaculture as a way of securing supplies. However, fish farming is not without its own problems.
Farmed fish are usually fed on fish oil and fishmeal (obtained by drying and grinding bones and offal left over from the processing of fish for human consumption together with fish unsuitable for human consumption), but there are problems associated with their use. Nitrogen and phosphorus present in the fishmeal can result in red tides, while the presence of phosphorus reduces the availability and absorption of dietary zinc in the fish being farmed. Furthermore, as aquaculture has expanded, the price of fishmeal has increased dramatically as demand has coincided with a decline in catches of the fish such as anchovies that are commonly used in fishmeal.
Professor Shuichi Satoh of the Tokyo University of Marine Science and Technology has therefore been working to develop a series of low- or non-fishmeal feeds based on corn or soya bean, which he has tested on red sea bream and yellowtail, the two major mariculture species in Japan. The red sea bream thrived on their new diet, growing bigger and faster than expected, even on a diet containing very little fishmeal [8]. In contrast, young yellowtail stopped growing after 40 days and exhibited green liver syndrome [9]. While taurine is present in fishmeal, it is missing from the plant-based alternatives, and unlike the red sea bream, the yellowtail cannot convert methionine or cysteine into taurine.
Satoh's team found that while the yellowtail grew normally if their feed contained 30% fishmeal, a taurine supplement was required if the feed contained only 20% fishmeal. Even then, there can be problems. Their biggest challenge has been to ensure that the fish eat the food they are given. Sometimes the yellowtail won't eat their new plant-based feed, or start to eat it but then spit it out again. Adding bonito peptides makes the feed taste more like fish, but also makes it more difficult to digest, so digestive enzymes have to be added as well; results so far are encouraging. While the team is also working on feeds based on poultry meal, pork, chicken meal and feather meal, they believe that plant-based mixtures will be the most environmentally friendly [8].
The background image is a Creative Commons licensed photograph of a school of Japanese yellowtail (Seriola quinqueradiata) in the south shore of Long-Dong Bay, a famous scuba diving site on the northeast coast of Taiwan, taken by Vincent C. Chen.
Reference(s)
- Tiedemann, F. and Gmelin, L. (1827) Einige neue Bestandtheile der Galle des Ochsen. Ann. Phys. Chem., 85(2), 326–337.
- Clauson, K.A., Shields, K.M., McQueen, C.E. and Persad, N. (2008) Safety issues associated with commercially available energy drinks. J. Am. Pharm. Assoc., 48(3), e55–63.
- Thimons, J.J. (2004) Examining taurine's role in ocular biochemistry and in CL wear. Contact Lens Spectrum, (4) .
- Thimons, J.J. (2004) Ocular health and next generation solutions. Understanding taurine and its possible role in ocular health. Contact Lens Spectrum, (8).
- Carrasco, S., Codoceo, R., Prieto, G., Lama, R. and Polanco, I. (1990) Effect of taurine supplements on growth, fat absorption and bile acid on cystic fibrosis. Acta Univ. Carol., Med., 36(1–4), 152–156.
- Rahman, M.M., Park, H.M., Kim, S.J., Go, H.K., Kim, G.B., Hong, C.U., Lee, Y.U., Kim, S.Z., Kim, J.S. and Kang, H,S. (2011) Taurine prevents hypertension and increases exercise capacity in rats with fructose-induced hypertension. Am. J. Hypertens., 24(5), 574–581.
- The State of the World Fisheries and Aquaculture, 2016. (Food and Agriculture Organization of the United Nations, Rome, 2016).
- Waycott, B. (2016) Could vegetarian fish be a thing of the future? Latest efforts from Japan. The Fish Site 1 September.
- Takagi, S., Murata, H., Goto, T., Ichiki, T., Munasinghe, D.M.S., Endo, M., Matsumoto, T., Sakurai, A., Hatate, H., Yoshida, T., Sakai, T., Yamashita, H., Ukawa, M. and Kuramoto, T. (2005) The green liver syndrome is caused by taurine deficiency in yellowtail, Seriola quinqueradiata fed diets without fishmeal. Aquacult. Sci., 53(3), 279–290.
13th September 2016, Lugdunin
There is currently an urgent need for new antibiotics to tackle the rise of antibiotic-resistant bacteria, such as Methicillin-resistant Staphylococcus aureus (MRSA). Antibiotic resistance is a growing cause for concern, with some experts warning of the possibility of up to 10 million unnecessary deaths worldwide by the year 2050. Most existing antibiotics have been obtained from soil bacteria and fungi but these are becoming more scarce and very few novel antibiotics have been developed since the 1980s. Now a group of scientists have discovered that a bacterial strain that is present in the human nostril produces an antibiotic, lugdunin (CHEBI:133127), which is capable of killing MRSA [1].Although usually harmless, S. aureus is present in the nasal passages of 30% of the population. However it can become invasive and cause deadly blood infections, especially in people with weakened immune systems. In their experiments, Dr. Bernhard Krismer, Alexander Zipperer and Professor Andreas Peschel from the Interfaculty Institute for Microbiology and Infection Medicine Tübingen, Germany observed that S. aureus is rarely found when another bacterium, Staphylococcus lugdunensis is present in the nose. As the two closely related microbes compete for survival in the nasal passages, S. lugdunensis secretes the antibiotic lugdunin that breaks down S. aureus, and thus eradicates its competitor. The team examined nasal swabs from 187 hospital patients; S. aureus was found in 5.9% in people who also carried S. lugdunensis, compared with 34.7% of those without it. In laboratory tests, lugdunin exhibited potent antimicrobial activity against a range of harmful gram-positive bacteria that are resistant to classical antibiotics. In mice, lugdunin applied topically was found to totally eradicate skin infections caused by S. aureus in both the upper and deeper layers of the flesh. In addition, bacteria obtained from these skin samples exhibited unchanged susceptibility to lugdunin, indicating that they had not developed resistance.
The next stage in the research will be to test the compound in humans. The researchers speculate that the drug might be able to be taken systemically, because it did not exhibit any signs of toxicity on a sample of human serum [1]. As S. lugdunensis can itself cause infections, there is also the potential for genetic modification of harmless bacteria so they can produce the antibiotic and be subsequently introduced to patients carrying S. aureus.
The background image is a Creative Commons licensed picture showing a hospital staff member wearing protective clothing to be used when in contact with patients with MRSA infection.
Reference(s)
- Zipperer, A., Konnerth, M.C., Laux, C., Berscheid, A., Janek, D., Weidenmaier, C., Burian, M., Schilling, N.A., Slavetinsky, C., Marschal, M., Willmann, M., Kalbacher, H., Schittek, B., BrËtz-Oesterhelt, H., Grond, S., Peschel, A. and Krismer, B. (2016). Human commensals producing a novel antibiotic impair pathogen colonization. Nature, 535, 511–516.

1st August 2016, Isobutyl butyrate
Compounds of interest in the fight against malaria have featured many times in ChEBI's Entity of the Month [1-6], and in recent years the number of cases of the disease has fallen markedly, due in no small part to the widespread adoption of insecticide-treated bednets (ITNs) and indoor residual spraying (IRS). Nevertheless, the disease continues to kill around a thousand people every day, of whom roughly 700 are children under the age of 5. More than 90% of the deaths occur in sub-Saharan Africa, where Plasmodium falciparum, the most dangerous species of the malaria parasite, predominates.While the ITNs/IRS strategy has resulted in a significant decline in the main vector of malaria transmission, the mosquito Anopheles gambiae, a shift to outdoor transmission by sympatric species, particularly An. arabiensis, has occurred. This has turned out to be a particularly difficult species to control, not least because it feeds both indoors and outdoors on a variety of vertebrate hosts.
Recently, research by a team of Swedish and Ethiopian scientists into how the mosquito selects its host has resulted in a possible new, simple and cheap way to prevent transmission of malaria or even Zika [7]. The study, led by Professor Rickard Ignell of the Department of Plant Protection Biology at the Swedish University of Agricultural Sciences, involved collecting mosquitoes from both indoors and outdoors in three villages in western Ethiopia and determining the source of their blood meals (human, cattle, goat, sheep or chicken) by ELISA testing. They found that indoors, An. arabiensis strongly prefers human blood, while outdoors, it avoids chickens, despite their high abundance, but feeds randomly on cattle, goats and sheep.
The mosquito locates its preferred host by detecting the profile of volatile compounds that is specific to each host. It is known that the detection of repellents (also known as non-host volatiles, NHVs) and attractants is used by the mosquito to aid in the selection of a host. The team therefore sampled the volatile compounds given off by both the host and the non-host species and determined the active compounds using gas chromatography (GC) combined with electroantennographic detection (EAD). These were then identified using GC combined with mass spectrometry (GC-MS).
Of the 11 GC-EAD active compounds detected from the chicken (the non-host), five were also found in one or more of the other hosts. Of the six that were only detected from the chicken, two could not be positively identified by comparison with commercially available synthetic standards. The four identified GC-EAD active compounds detected only from the chicken were isobutyl butyrate (CHEBI:87683), hexadecane, naphthalene, and trans-limonene 1,2-epoxide.
In field trials, volunteers in 11 similar-sized thatched houses in an Ethiopian village slept under an untreated bednet with a CDC suction trap (with the light bulb removed) hung at the foot of the bed, about 1 m above the ground. Nine of the traps were baited with different GC-EAD active compounds from the host (cattle, goats and sheep) and non-host (chicken) species; a control trap had only solvent, while in one house, a caged chicken surrounded by a fine mesh screen (to "prevent chicken-mosquito interactions" – chickens are known to eat mosquitoes) was suspended near an unbaited trap. The experiment was repeated for a further 10 nights, with the traps/chicken being rotated around the 11 houses so as to minimise any possible location bias in the results.
The researchers found that traps baited with a live, caged chicken, or with the chicken-specific compounds, caught significantly fewer mosquitoes than control traps. Traps baited with three of the "generic" compounds tested (limonene, cis-limonene 1,2-epoxide and β-myrcene) also caught fewer mosquitoes than the control, while the other "generic" compounds tested, sulcatone and nonanal, had no effect on the number of An. arabiensis caught.
The team believe that, in combination with established control programmes, the NHVs they identified could be employed to afford significant protection to people at risk of contracting mosquito-vectored diseases. They now intend to determine the effectiveness and duration of action of an appropriately-formulated repellent product. Isobutyl butyrate in particular is likely to be included in any such product, as it simple and cheap to manufacture, has a pleasant, fruity odour (a characteristic property of most volatile carboxylic esters), and has been widely used as a food flavour ingredient and in perfumery for many years. With resistance to insecticides becoming an increasing problem, the development of new methods and products for the control of malaria will be welcome news.
The background image is a Creative Commons licensed photograph of a Light Sussex hen at Collingwood Children's Farm, Melbourne, Australia.
Reference(s)
- Entity of the Month 1st March 2016, WLL-vs.
- Entity of the Month 4th March 2013, Gedunin.
- Entity of the Month 6th February 2012, (+)-Artemisinin.
- Entity of the Month 4th November 2009, Artemether.
- Entity of the Month 7th October 2009, Chloroquine.
- Entity of the Month 26th October 2005, Quinacrine.
- Jaleta K.T., Hill, S.R., Birgersson, G., Tekie, H., and Ignell, R. (2016) Chicken volatiles repel host-seeking malaria mosquitoes. Malar. J., 15, 354.

1st July 2016, Phenylephrine
While an effective cure for the common cold remains elusive, there is a large market for over the counter cold remedies containing decongestants, that provide some relief from the symptoms. Until 2008 it was possible to walk into a shop on the high street and purchase a cold relief medicine that contained the active decongestant pseudoephedrine. This drug has been used for several decades and has proved to be an effective decongestant, but in 2008 it was placed under tight controls when it became apparent that pseudoephedrine was being used for production of the illicit drug methamphetamine (otherwise known as crystal meth). Since then, pseudoephedrine has been largely absent from shop shelves in the UK, the US and elsewhere. Currently, products containing this drug are only available either through obtaining a prescription, or by requesting a packet from behind the counter of a pharmacy store. Drug companies have responded to the regulations by replacing pseudoephedrine with phenylephrine (CHEBI:8093), a drug already approved by the US Food and Drug Administration (FDA), but known to be significantly less effective.Since then the actual efficacy of phenylephrine has been called into question. In 2006, cold expert Ronald Eccles at Cardiff University, UK, conducted a review of phenylephrine research and reported that he could not find any evidence that the drug is effective as a decongestant when taken orally [1]. Subsequent studies showed that FDA-approved 10 mg doses of phenylephrine were no more effective than a placebo [2] while a conflicting review indicated that the drug had a small but real effect on nasal airway resistance [3]. As a result, the FDA requested that large, well-constructed, dose-ranging trials be conducted. The study, carried out at the University of Florida, US, involved 539 adults, lasted one week and failed to find a dose of phenylephrine within the 10 mg to 40 mg range that was significantly more effective than a placebo in relieving nasal congestion [4]. Consequently, the researchers have submitted a citizens' petition to the FDA calling for the removal of oral phenylephrine from the over the counter market.
The background image is a Creative Commons licensed picture showing the interior of Shaw's Pharmacy, Seattle, Washington (from 1907).
Reference(s)
- Eccles, R. (2007). Substitution of phenylephrine for pseudoephedrine as a nasal decongeststant. An illogical way to control methamphetamine abuse. Brit. J. Clin. Pharmaco., 63, 10–14.
- Hendeles, L. and Hatton, R.C. (2006). Oral phenylephrine: An ineffective replacement for pseudoephedrine? J. Allergy Clin. Immun., 118, 279–280.
- Kollar, C., Schneider, H., Waksman, J. and Krusinska, E. (2007). Meta-analysis of the efficacy of a single dose of phenylephrine 10 mg compared with placebo in adults with acute nasal congestion due to the common cold. Clin. Ther., 29, 1057–1070.
- Meltzer, E.O, Ratner, P.H. and McGraw, T. (2015). Oral phenylephrine HCl for nasal congestion in seasonal allergic rhinitis: A randomized, open-label, placebo-controlled study. J. Allergy Clin. Immun. Pract., 3, 702–708.
1st June 2016, (R)-Amygdalin
Wheat is the predominant cereal crop in the world and vermin infestation remains a serious issue regarding its storage and field cultivation. Many cereal crops are ill-protected against insect pests during storage periods. While pesticides treatments are undoubtedly effective, there are increasing concerns about their environmental and ecological impact. A team of scientists led by Wendelin Stark at ETH Zurich have developed a novel approach that mimics the defence system of bitter almonds and other members of the Prunus family [1]. Their seeds contain the cyanogenic glycoside amygdalin (CHEBI:17019) together with a β-glucosidase enzyme. When the seed is attacked, the enzyme combines with the amygdalin resulting in the release of toxic hydrogen cyanide (HCN), thus preventing the seed from being eaten.In the laboratory, the research team coated wheat seeds with several layers of polylactic acid (PLA), a substance that is both harmless and biodegradable. The innermost layer contained the enzyme. On top of this lay a further layer of PLA, followed by two layers in which amygdalin was embedded. When insect larvae chewed through these layers, the amygdalin was released, followed by the enzyme resulting in release of HCN. In collaboration with the Julius Kühn Institute in Berlin, the researchers tested the efficacy of their treatment on a variety of cereal pests. The defence system proved to be very effective against the larvae of the mealworm (Tenebrio molitor), the Indian mealmoth (Plodia interpunctella) and the lesser grain borer (Rhizopertha dominica, a beetle that causes considerable damage to wheat stores worldwide). Although the treatment did not prove fatal to any of the tested insect pest species, significant reduction of their reproduction and infestation rate was observed.
In a previous study, mandelonitrile was used as the HCN precursor, but this method proved to be impractical due to a very low seed germination rate [2]. Field studies with the new system showed that although the coated grains did germinate at a slightly reduced rate compared with the uncoated ones, the plants were able to recover and the effect became negligible during later growth stages. In addition, the new method is straightforward and cost-effective and hence has potential as an alternative to conventional pesticide treatments.
The background image is a Creative Commons licensed picture showing a lesser grain borer feeding on wheat.
Reference(s)
- Mora, C.A., Halter, J.G., Adler, C., Hund, A., Anders, H., Yu, K. and Stark, W.J. (2016). Application of the Prunus spp. cyanide seed defense system onto wheat: reduced insect feeding and field growth tests. J. Agric. Food Chem., 64, 3501–3507.
- Halter, J.G., Chen, W.D., Hild, N., Mora, C.A., Stoessel, P.R., Koehler, F.M., Grass, R.N. and Stark, W.J. (2014). Induced cyanogenesis from hydroxynitrile lyase and mandelonitrile on wheat with polylactic acid multilayer-coating produces self-defending seeds. J. Mater. Chem. A, 2, 853–858.
1st May 2016, Phorbol
An evergreen member of the spurge (Euphorbiaceae) family, the manchineel tree (Hippomane mancinella) is found along beaches and in swamps in tropical southern North America, Central America, and the West Indies, where it is often cultivated to provide windbreaks. While it can survive in a stunted form, only a few inches high, on windward cliffs, in more favourable conditions it can grow to around 15 m (50 ft) tall. The yellow-green or yellowish fruits of the tree, which look and smell like small apples, give the tree its alternative name – the beach apple. Guinness World Records classes it as the most dangerous tree in the world [1].When any part of the tree is cut or broken, a milky-white sap or latex is exuded that is intensely irritating. Contact of the sap with the skin causes blistering and allergic dermatitis. Simply standing under a manchineel during rain can result in skin blistering due the raindrops dissolving small amounts of exuded sap as they run off of the leaves [2]. Contact of the sap with the eyes is also extremely unpleasant and can cause acute keratoconjunctivitis [3]; even smoke from the burning wood of a manchineel can result in eye damage.
The apple-like fruits of the tree are also dangerous. A vivid description of a holiday misadventure in the BMJ describes how the apple tastes "pleasantly sweet" at first, but then there is an odd peppery feeling that slowly becomes a "burning, tearing sensation and tightness of the throat". After a couple of hours, the author could "barely swallow solid food because of the excruciating pain and the feeling of a huge obstructing pharyngeal lump." The symptoms gradually declined over the next eight hours; the pain was slightly diminished by drinking milk, while most alcoholic beverages made it worse [4].
The numerous compounds responsible for the irritant properties of manchineel sap include several ester and orthoester derivatives of phorbol (CHEBI:8116) [5]. Phorbol itself was first isolated in 1934 as a hydrolysis product of croton oil (obtained from the seeds of the purging croton plant, Croton tiglium and formerly used as a drastic cathartic and counterirritant) [6]. A tetracyclic diterpenoid with a skeleton based on tigliane, it took over 30 years before its structure was finally settled [7,8].
The biological properties of various esters of phorbol, notably as tumour promoters and activators of protein kinase C, have attracted the attention of researchers for many years. Of particular interest has been phorbol 13-acetate 12-myristate, currently in phase II clinical trials for the treatment of acute myeloid leukaemia. The complex structure of phorbol, containing four fused rings and eight chiral centres, has meant that to date, the preparation of phorbol derivatives has depended on semisynthesis – the synthetic modification of naturally-sourced phorbol or phorbol-related compounds. A downside of this technique is that, with a restricted structure as the starting material, it can often be impossible to prepare certain desired analogues. In principle, this problem could be overcome by a total synthesis approach, but until recently only two total syntheses and two formal syntheses of phorbol had been reported, and they involved between 40 and 52 separate steps, making them impractical on a preparative-scale.
In a remarkable demonstration of the power of synthetic organic chemistry, a team led by Professor Phil Baran at the Scripps Research Institute in La Jolla, California, has recently reported a new synthesis of (+)-phorbol from the readily-available monoterpene (+)-car-3-ene using a route that was designed specifically to permit the preparation of phorbol analogues containing unique placements of oxygen atoms that were previously inaccessible [9]. The elegance of the team's strategy is highlighted not only by it being dramatically shorter than all previous syntheses – just 19 steps – but by the fact that no new chemistry was required to achieve it; all of the reactions employed have been in the synthetic chemist's arsenal for decades.
The background image is a Creative Commons licensed photograph showing the fruit of the manchineel tree.
Reference(s)
- Guinness World Records. Most Dangerous Tree.
- Nellis, D.W. Poisonous plants and animals of Florida and the Caribbean. Pineapple Press, Inc., Sarasoto, Florida. (1997) p. 173.
- Pitts, J.F., Barker, N.H., Gibbons, D.C. and Jay, J.L. (1993) Manchineel keratoconjunctivitis. Br. J. Ophthalmol., 77(5), 284–288.
- Strickland, N.H., Glennie, A. and Sanderson, H (2000) Eating a manchineel "beach apple". BMJ, 321(7258), 428.
- Adolf, W. and Hecker, E. (1984) On the active principles of the spurge family, X. Skin irritants, cocarcinogens, and cryptic cocarcinogens from the latex of the manchineel tree. J. Nat. Prod., 47(3), 482–496.
- Flaschenträger, B. and v. Wolffersdorff, R. (1934) Über den Giftstoff des Crotonöles. 1. Die Säuren des Crotonöles. Helv. Chim. Acta, 17(1), 1444–1452.
- Elix, J.A., Roffey, P. and Sargent, M.V. (1967) The conversion of tri-O-methylsolorinic acid into tetra-O-methylaverythrin. Tetrahedron Lett., 8(33), 3161–3163.
- Pettersen, R.C., Ferguson, G., Crombie, L., Games, M.L. and Pointer, D.J. (1967) The structure and stereochemistry of phorbol, diterpene parent of co-carcinogens of croton oil. Chem. Commun. (London), (14), 716–717.
- Kawamura, S., Chu, H., Felding, J, and Baran, P.S. (2016) Nineteen-step total synthesis of (+)-phorbol. Nature, 532(7597), 90–93.
1st April 2016, Tarocin A2
Methicillin-resistant Staphylococcus aureus, commonly known as MRSA, is a major cause of hospital-acquired infections, which can often result in death. These bacteria have developed resistance to the most commonly employed broad-spectrum β-lactam antibiotics, such as penicillins, cephalosporins and even carbapenems. While strict enforcement of sanitation procedures, particularly thorough hand washing and the use of alcohol rubs by all medical personnel before and after contact with each patient, has dramatically reduced infection rates in hospitals, there is nevertheless an urgent need for new treatment options. One approach involves the use of existing β-lactam antibiotics in combination with compounds that can hamper the bacterial resistance and hence restore their efficacy.One such study has been carried out by a team led by Christopher Tan, Director of Infectious Diseases at Merck Research Laboratories in New Jersey [1]. Here, they focused on compounds that inhibit the biosynthesis of teichoic acid, a major structural component of the bacterial cell wall that is thought to protect MRSA against the antimicrobial effects of β-lactams.
The researchers identified two synthetic compounds, named tarocin A and tarocin B, that were found to disrupt teichoic acid production at an early stage. When administered alone, these compounds had no toxic effects on either MRSA or human cells. When paired with common β-lactam antibiotics, however, various clinical strains of resistant bacteria were killed. Next, a study with mice infected with MRSA was carried out using a more potent analogue of tarocin A, known as tarocin A2 (CHEBI:131615) in combination with the β-lactam antibiotic dicloxacillin. The team found that mice treated with both drugs had reduced levels of infection after 24 hours, while those given one drug or the other showed no signs of improvement. Moreover, the drug combination did not appear to be toxic to the mice. The next stage of the research will be to carry out tests in human patients.
The background image is a Creative Commons licensed photograph depicting the human neutrophil ingesting MRSA.
Reference(s)
- Lee, S.H., Wang, H., Labroli, M., Koseoglu, S., Zuck, P., Mayhood, T., Gill, C., Mann, P., Sher, X., Ha, S., Yang, S.W., Mandal, M., Yang, C., Liang, L., Tan, Z., Tawa, P., Hou, Y., Kuvelkar, R., DeVito, K., Wen, X., Xiao, J., Batchlett, M., Balibar, C.J., Liu, J., Xiao, J., Murgolo, N., Garlisi, C.G., Sheth, P.R., Flattery, A., Su, J., Tan, C. and Roemer, T. (2016). TarO-specific inhibitors of wall teichoic acid biosynthesis restore β-lactam efficacy against methicillin-resistant staphylococci. Sci. Transl. Med., 8, 329–332.

1st March 2016, WLL-vs
Funding for the prevention and treatment of malaria has increased more than 10-fold since the year 2000, with dramatic results. Increased prevention and control measures, including the provision and use of bed nets impregnated with long-lasting (2-3 years) insecticides to protect people from mosquito bites at night, together with indoor spraying with insecticides, has resulted in a 37% reduction in new malaria cases and a 60% reduction in malaria mortality rates [1].Despite these advances, nearly half of the world's population is still at risk of malaria. Last year, there were over 200 million cases of malaria and an estimated 438,000 malaria deaths; around 70% of the latter were of children under 5. More than 90% of malarial deaths occur in sub-Saharan Africa, which is a stronghold of the most dangerous species of malaria parasite, Plasmodium falciparum [1].
Resistance of the malaria parasite to (+)-artemisinin – the core compound in the recommended combination treatments for uncomplicated malaria – has already been found in 5 countries in Southeast Asia and seems certain to spread, since most treatments do not prevent transmission of the disease. Consequently, it seems certain that the current tools and treatments will not be capable of eliminating malaria and that new antimalarial agents will be required.
Found in all eukaryotes and archaea, proteasomes are protein complexes whose main function is to break down unwanted damaged or misfolded proteins. They form part of the mechanism by which cells regulate the cell cycle, gene expression, and response to oxidative stress. In recent years, compounds that inhibit the action of proteasomes have become an attractive target for new antimalarial drugs.
Unlike current antimalarial drugs, proteasome inhibitors have been shown to be toxic to all nine stages of the complex life cycle of Plasmodium falciparum [2,3]. Combination of a suitable proteasome inhibitor with artemisin could therefore help to reduce the spread of malarial drug resistance. Unfortunately, most of the proteasome inhibitors tested so far also inhibit mammalian proteasomes as well as Plasmodium proteasomes, resulting in toxic side effects in humans and preventing their use as antimalarial drugs.
Now, a team led by Prof. Matthew Bogyo at Stanford University School of Medicine have screened a 228-member library of tetradecapeptides to find amino acid sequences degraded by parasite proteasomes but not human ones. They used the results of their screens to design selective inhibitors. The structures of the new inhibitors when bound to the parasite proteasome were subsequently determined using cryo-electron microscopy by Paula da Fonseca at the MRC Laboratory of Molecular Biology in Cambridge, UK, enabling the inhibitor design to be further optimised [4].
The most successful compound to emerge from the work was termed WLL-vs (CHEBI:91210), as it is a vinyl sulfone (vs) derived from the tripeptide tryptophyl-leucyl-leucine, whose designation using the 1-letter amino acid code is WLL. Treatment of mice with a single dose of WLL-vs three days after they had been infected with one million parasites resulted in virtual elimination of the parasite with no toxic effects being observed. WLL-vs was found to kill both artemisinin-sensitive and -resistant malaria parasites [4].
While WLL-vs still has many regulatory hurdles to cross before it can be considered for use as a component of a new treatment for malaria, the work of Bogyo and his fellow researchers has shown that the Plasmodium proteasome is a feasible target for future antimalarial drugs. Furthermore, the group's strategy could be more widely applicable for finding proteasome-specific lead compounds for other parasitic diseases, such as toxoplasmosis and leishmaniasis.
The background image is a Creative Commons licensed photograph of a Yugoslavian postage stamp issued on April 7, 1962 to publicise the World Health Organization's campaign to eradicate malaria. The inscription reads: 'The world united against malaria'.
Reference(s)
- World Malaria Report 2015. (World Health Organisation, Geneva, 2015).
- Kreidenweiss, A., Kremsner, P.G. and Mordmüller, B. (2008) Comprehensive study of proteasome inhibitors against Plasmodium falciparum laboratory strains and field isolates from Gabon. Malar. J., 7(10), 187.
- Czesny, B., Goshu, S., Cook, J.L. and Williamson, K.C. (2009) The proteasome inhibitor epoxomicin has potent Plasmodium falciparum gametocytocidal activity. Antimicrob. Agents Chemother., 53(10), 4080–4085.
- Li, H., O'Donoghue, A.J., van der Linden, W.A., Xie, S.C., Yoo, E., Foe, I.T., Tilley, L., Craik, C.S., da Fonseca, P.C. and Bogyo, M. (2016) Structure- and function-based design of Plasmodium-selective proteasome inhibitors. Nature, 530(7589), 233–236.

1st February 2016, S-Propyl propanethiosulfinate
Many plants are famed for their putrid smells, and it has been known for decades that unpleasant sulphurous odours are released when soil is disturbed around the roots of some plants. It has always been assumed that these odours are released passively as a result of tissue damage. Now a team of researchers led by Dr. Rabi Musah at the University at Albany in New York has discovered a previously unknown defence mechanism in plants: roots that actively release a foul odour when they sense the touch of a potential predator [1].Studies were performed using Mimosa pudica, a perennial shrub native to Brazil but now widely distributed throughout the world. Its roots are known to emit a cocktail of small organic and inorganic sulfur compounds into the environment, but with no detectable odour. However, when soil around the roots is dislodged or when seedling roots are touched, a distinctive odour is detected. By using high resolution mass spectrometry techniques, the researchers were able to attribute the odour to the emission of increased amounts of S-propyl propanethiosulfinate (CHEBI:91021), propanesulfenic acid, 2-aminothiophenol and phenothiazine. Moreover, the team also observed that the roots seemed to distinguish between different kinds of touch. While touching the roots with soil or human skin elicited a strong odour, agitating the roots with other materials such as glass or metal did not induce a similar response. Finally, by growing seedlings of M. pudica in a sterile environment, the team were able to ascertain that emission of the organosulfur volatiles occurred exclusively from the roots and not the aerial parts of the plant. The team have also found that at least six other Mimosa species produce the same smell and now plan to study plants in the closely related Acacia genus.
The background image is a Creative Commons licensed picture showing the purple flower head of Mimosa pudica.
Reference(s)
- Musah, R.A., Lesiak, A.D., Maron, M.J., Cody, R.B., Edwards, D., Fowble, K.L., Dane, A.J. and Long, M.C. (2015). Mechanosensitivity below ground: touch-sensitive smell-producing roots in the "shy plant," Mimosa pudica L. Plant Physiol, Dec 9. pii: pp.01705.2015.[Epub ahead of print].
1st January 2016, Naloxone
First patented in 1961 by the Japanese pharmaceutical company Sankyo, the opioid receptor antagonist naloxone CHEBI:7459 has been used as an antidote to opioid overdose for over forty years and is on the World Health Organization's Model List of medicines considered to be essential for a basic health care system. More recently, however, it has made news due to its role in research that enabled a woman to feel pain for the first time in her life.Congenital analgesia arises from an extremely rare genetic mutation that results in a lack of the Nav1.7 ion channels that transport sodium across sensory nerves. Without these channels, nerve cells cannot communicate pain, so people who have the mutation are born unable to feel pain. As a result, they do not know when they have been injured: babies with the condition frequently chew their lips, fingers and toes until they bleed, and as they grow into toddlers and become more mobile, so the risk of serious injury from knocks, falls, scalds and burns increases.
Since people who lack the Nav1.7 channels cannot feel pain, it seemed logical that compounds that blocked these channels could act as painkillers in people who don't have the disorder. Consequently, the discovery of Nav1.7 channel blockers became an important target for drug research. Although some successful compounds were found, none resulted in the total pain loss experienced by those who naturally lack the Nav1.7 channels.
To investigate this phenomenon, Prof. John Wood and colleagues at University College London studied the nerves in mice that had been genetically modified to lack the Nav1.7 channels [1]. They found that the Nav1.7 deletion resulted in a large increase in the expression of genes responsible for the enkephalin peptides, the body's natural painkillers. Wood's team reasoned that it was possible that people who lacked the Nav1.7 channels could also be producing more enkephalins, resulting in total pain loss. If so, then treating a sufferer of congenital analgesia with a compound that blocks the action of the opioid peptides, such as naloxone, could reverse the disorder.
After successful experiments on mice, the researchers tried naloxone with a 39-year-old woman with congenital analgesia. After she was given the drug, she was able to feel the pain caused by the heating effect of a laser – the first time in her life she had felt pain.
The background image is a detail from a Creative Commons licensed photograph of a 16th Century shield depicting the legendary Roman hero Gaius Mucius Scaevola. Gaius Mucius was a Roman youth who was captured after he failed in an attempt to kill the Etruscan king Lars Porsena during the Roman - Etruscan war c. 508 BC. Mucius declared that he was just the first of 300 noble youths who would attempt to kill the king, one after another until they succeeded. To demonstrate his courage, Mucius thrust his hand into an altar fire, holding it there until it was consumed. Astonished and impressed by this deed and afraid of another attempt on his life, the king released Mucius and made peace with Rome. Mucius was rewarded with a grant of farming land and given the name Scaevola, meaning 'left handed'.
Reference(s)
- Minett, M.S., Pereira, V., Sikandar, S., Matsuyama, A., Lolignier, S, Kanellopoulos, A.H., Mancini F., Iannetti, G.D., Bogdanov, Y.D., Santana-Varela, S., Millet, Q., Baskozos, G., MacAllister, R., Cox J.J., Zhao, J., and Wood, J.N. (2015) Endogenous opioids contribute to insensitivity to pain in humans and mice lacking sodium channel Nav1.7. Nature Commun., 6, 8967.
2015
1st December 2015, Disulfiram
The battle against human immunodeficiency virus (HIV) infection is still ongoing, and scientists are continuing the search for an effective cure for the disease. Use of suppressive anti-retroviral therapy (ART) treatment can eradicate HIV from the bloodstream, but the virus can hide in a dormant state elsewhere in the body. To prevent the virus re-emerging, people infected with HIV have to remain on these drugs for the rest of their lives. Although some headway has been made with activating latent HIV, the drugs trialled have had too many toxic side effects to be a viable option.Now a group of scientists led by Professor Sharon Lewin at the University of Melbourne, Australia believe they may have found a non-toxic alternative for activating latent HIV [1]. The compound in question is the generic drug disulfiram (CHEBI:4659, brand name Antabuse), which has been used over several decades for alcohol aversion therapy.The team tested the drug on 30 patients infected with HIV over a period of three days and found an increase in HIV gene expression in all of the volunteers, a sign that the dormant virus may have been 'woken up'. If bigger trials support ART findings, this could be a vital step towards finding a cure for HIV. The next phase is to find a second drug that can eliminate the re-awakened virus from the body. While current ART treatment can prevent infected cells from multiplying, it cannot destroy them. Clearly, there is still some way to go before scientists are able to ultimately eradicate this very smart virus.
The background image is a detail from a Creative Commons licensed picture showing a human white blood cell being attacked by the HIV virus. The file comes from Wellcome Images, a website operated by the Wellcome Trust, a global charitable foundation based in the UK, and is a copyrighted work available under Creative Commons Attribution only licence CC BY 4.0.
Reference(s)
- Elliott, J.H., McMahon, J.H., Chang, C.C., Lee, S.A., Hartogensis, W., Bumpus, N., Savic, R., Roney, J., Hoh, R., Solomon, A., Platak, M., Gorelick, R.J., Lifson, J., Bacchetti, P., Deeks, S.G. and Lewin, S.R. (2015). Short-term administration of disulfiram for reversal of latent HIV infection: a phase 2 dose-escalation study The Lancet HIV, doi:10.1016/S2352-3018(15)00226-X, published online 16 November 2015.
1st November 2015, trans-Resveratrol
trans-Resveratrol (trans-RV, CHEBI:45713) is found in peanuts, grapes, red wine, and some berries, but was first isolated from the roots of the medicinal plant Veratrum grandiflorum (white hellebore) in 1940 by Mitio Takaoka in Hokkaido Imperial University [1]. More than twenty years passed before the compound received further attention: in 1963, trans-RV was found in the roots of Japanese knotweed by a group of Japanese researchers studying various plants used in traditional Chinese and Japanese herbal remedies [2]. trans-RV was then neglected for a further two decades.In 1992, Evan Siemann and Leroy Creasy reported finding trans-RV in red wine and thought that it might explain the "French Paradox" – that the French have a relatively low rate of coronary heart disease despite having a high rate of smoking and consuming a diet that is relatively high in saturated fats [3]. It is known that calorie restriction can extend the lifespan of a number of species, including yeast, worms, flies, fish, rats, and mice [4]. In yeast, calorie restriction stimulates the activity of the Silent information regulator 2 protein (also known as Sir2 or sirtuin). In 2002, David Sinclair and collaborators found that providing yeast with trans-RV in the absence of calorie restriction activated the sirtuin protein and extend the replicative lifespan of yeast by 70% [5]. Feeding with trans-RV was also found to extend the lifespan of animal species, including worms (Caenorhabditis elegans), fruit flies (Drosophila melanogaster), and a vertebrate fish (Nothobranchius furzeri) and to extend the (normally shortened) lifespan of mice on a high-calorie diet to one that was similar to that of mice on a standard diet [6-8]. Despite many years of investigation, however, there is no evidence that incorporating trans-RV in the diet can increase human lifespan – the concentrations of resveratrol required to increase human sirtuin activity are considerably higher than those that have been measured in human plasma after oral consumption of trans-RV.
The discovery that in a mouse model of Alzheimer's disease (AD), calorie restriction limited the production and deposition of neurotoxic β-amyloid peptide in the brain [9] and the subsequent finding that trans-RV promotes the AMP-activated protein kinase (AMPK) dependent clearance of β-amyloid peptide in the brain of an AD mouse model [10] has prompted further investigation into the possibility of using trans-RV to reduce or treat cognitive decline. Recently, Prof. Raymond Scott Turner of Georgetown University in Washington has reported the results of a collaborative study in which 119 people with mild to moderate symptoms of AD were split into two groups, given either a placebo or 1 g of trans-RV (equivalent to the amount found in over 100 bottles of red wine!) twice a day for a year [11].
Although the main purpose of the study was to determine the safety and tolerability of trans-RV , the group also observed that over the course of the study, the level of β-amyloid peptide in the blood of the placebo group declined as the AD progressed – thought to be a result of the β-amyloid peptide being deposited in their brains. However, in the blood of the group taking trans-RV, there was little or no change in the β-amyloid peptide levels. There are many questions about AD that are still unanswered, including the role of β-amyloid deposits in the brain – are they a cause of the condition or merely a symptom? Whether trans-RV will prove to be of use in tackling cognitive decline remains to be seen.
The background image is a detail from a Creative Commons licensed picture showing different parts of the peanut plant, Arachis hypogaea (a rich source of trans-resveratrol), published in Köhler's Medicinal Plants (1887).
Reference(s)
- Takaoka, M. (1940) Phenolic substances of white hellebore (Veratrum grandiflorum Loes. fil.) J. Faculty Sci., Hokkaido Imp. Univ., 3(Ser. III), 1–16.
- Nonomura, S., Kanagawa, H. and Makimoto, A. (1963) Chemical constituents of polygonaceous plants. I. Studies on the components of ko-jo-kon (Polygonum cuspidatum Sieb. et Zucc.). Yakugaku Zasshi, 83, 988â990.
- Siemann, E. and Creasy, L. (1992) Concentration of the phytoalexin resveratrol in wine. Am. J. Enol. Vitic., 43(1), 49–52.
- Heilbronn, L.K. and Ravussin, E. (2003) Calorie restriction and aging: review of the literature and implications for studies in humans. Am. J. Clin. Nutr., 78(3), 361–369.
- Howitz, K.T., Bitterman, K.J., Cohen, H.Y., Lamming, D.W., Lavu, S., Wood, J.G., Zipkin, R.E., Chung, P., Kisielewski, A., Zhang, L.-L., Scherer, B. and Sinclair, D.A. (2003) Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan.Nature, 425(6954), 191–196.
- Wood, J.G., Rogina, B., Lavu, S., Howitz, K., Helfand, S.L., Tatar, M. and Sinclair, D. (2004) Sirtuin activators mimic caloric restriction and delay ageing in metazoans. Nature, 430(7000), 686–689.
- Valenzano, D.R., Terzibasi, E., Genade, T., Cattaneo, A., Domenici, L. and Cellerino, A. (2006) Resveratrol prolongs lifespan and retards the onset of age-related markers in a short-lived vertebrate. Curr. Biol., 16(3), 296–300.
- Baur, J.A., Pearson, K.J., Price, N.L., Jamieson, H.A., Lerin, C., Kalra, A., Prabhu, V.V., Allard, J.S., Lopez-Lluch, G., Lewis, K., Pistell, P.J., Poosala, S., Becker, K.G., Boss, O., Gwinn, D., Wang, M., Ramaswamy, S., Fishbein, K.W., Spencer, R.G., Lakatta, E.G., Le Couteur, D., Shaw, R.J., Navas, P., Puigserver, P., Ingram, D.K., de Cabo, R. and Sinclair, D.A. (2006) Resveratrol improves health and survival of mice on a high-calorie diet. Nature, 444(7117), 337–342.
- Wang, J., Ho, L., Qin, W., Rocher, A.B., Seror, I., Humala, N., Maniar, K., Dolios, G., Wang, R., Hof, P.R. and Pasinetti, G.M. (2005) Caloric restriction attenuates β-amyloid neuropathology in a mouse model of Alzheimer's disease. FASEB J, 19(6), 659–661.
- Vingtdeux, V., Giliberto, L., Zhao, H., Chandakkar, P., Wu, Q., Simon, J.E., Janle, E.M., Lobo, J., Ferruzzi, M.G., Davies, P. and Marambaud, P. (2010) AMP-activated protein kinase signaling activation by resveratrol modulates amyloid-beta peptide metabolism. J. Biol. Chem., 285(12), 9100–9113.
- Turner, R.S., Thomas, R.G., Craft, S., van Dyck, C.H., Mintzer, J., Reynolds, B.A., Brewer, J.B., Rissman, R.A., Raman, R. and Aisen, P.S. (2015) A randomized, double-blind, placebo-controlled trial of resveratrol for Alzheimer disease. Neurology, 85(16), 1383–1391.

1st October 2015, Hydrogen peroxide
In our Entity of the Month article for March 2012, we highlighted an unusual example of an insect species utilising alcohol for self-medication to ward off a blood-borne parasite. Self-medication in insects involves selectively consuming substances that are otherwise harmful in order to fight infections or deter predators. Although interest has subsequently heightened, conclusive evidence of this behaviour pattern in insects has proved to be elusive. Now, a team led by Nick Bos at the University of Helsinki, Finland, has demonstrated that ants of the Formica fusca species actively choose to ingest hydrogen peroxide (H2O2, CHEBI:16240) when infected by a dangerous fungal disease and are more likely to survive as a result [1].Firstly, the researchers demonstrated that hydrogen peroxide is actually harmful to the ants by feeding them with either a simple honey-based solution, or the same solution containing hydrogen peroxide. It was shown that healthy ants receiving the spiked diet had a mortality rate of around 20%, compared with around 5% for those fed with the control solution. Next, the feeding experiments were performed using ants infected with the parasitic fungus Beauveria bassiana. Here, the pattern was reversed with mortality rates of the infected ants falling from around 60% for those on the ordinary diet to 45% in those receiving food laced with hydrogen peroxide. When the ants were offered a choice, the healthy ants tended to avoid the spiked food while those carrying the infection chose to consume increased amounts of the solution containing hydrogen peroxide, and also seemed to choose their dosage carefully. They tended to avoid the supplemented food when the concentration of hydrogen peroxide was too high. Although the drop in mortality rate may seem small percentage-wise, the extra surviving individuals can be crucial for the viability of large ant colonies.
Hydrogen peroxide is a common precursor to reactive oxygen species (ROS), chemicals which are harmful to the host organism but also assist in defending that organism against invading pathogens. The team propose that under natural conditions, the ants might adjust their intake of ROS either by seeking out plants that are rich in ROS or by increasing or decreasing their predation of aphids, which in turn feed on the ROS-rich plants [2]. The next step for the team will be to investigate whether ants obtain this medicine solely from their aphid symbionts, or whether they have access to other sources of ROS.
The background image is a detail from a Creative Commons licensed image depicting a Formica fusca specimen.
Reference(s)
- Bos, N., Sundstrom, L., Fuchs, S. and Freitak, D. (2015). Ants medicate to fight disease. Evolution, doi:10.1111/evo.12752, published online 23 July 2015.
- Offenberg, J. (2001). Balancing between mutualism and exploitation: the symbiotic interaction between Lasius ants and aphids. Behav. Ecol. Sociobiol., 49, 304–310.
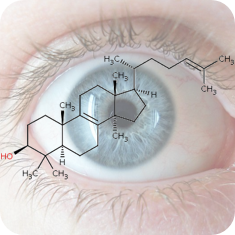
1st September 2015, Lanosterol
The lens of the eye is made up of many millions of thin fibrous cells that contain water-soluble structural proteins known as crystallins. These proteins are among the densest of human tissues and their main function is to increase the refractive index of the lens while not obstructing light. Despite their name, crystallins have been selected by evolution for their tendency not to form crystals – the presence of small crystals or aggregates within the lens would scatter light, so making the lens opaque.There are three types of crystallin: α (the most common), β, and γ. α-Crystallins are composed of two similar types of protein chain which associate into large spherical complexes, containing around 40 chains, that repel each other and so spread themselves throughout the lens cells. β-Crystallins also form oligomeric complexes, usually containing either two or six copies of the chain. There are a number of similar β-crystallins, which can "mix and match" to give a large number of different types of oligomers. γ-Crystallins, in contrast, are monomeric, and appear to act as a weak glue that binds the α-crystallins gently together [1].
As we age, crystallins may unfold or be oxidised or otherwise damaged. As the damage builds up, so opaque clumps are formed which scatter light, leading to a cloudiness in the lens, eventually resulting in a cataract. Cataracts are the cause of a third of all visual impairment in the world and half of all blindness; they are the most common cause of vision loss in people over the age of 40 [2]. Treatment is by surgery: a small incision is made in the eye to allow the affected lens to be removed and replaced by a plastic lens, called an intraocular implant or intraocular lens. It is one of the most common surgical procedures – more than 300,000 such operations are performed each year in the UK alone [3].
Recent work by a team of Chinese researchers suggests that an alternative treatment strategy may be possible [4]. The team studied the DNA of two families with a genetic predisposition to cataracts and found two mutations in the LSS gene, which codes for lanosterol synthase, an enzyme that catalyses the conversion of (S)-2,3-epoxysqualene to lanosterol (CHEBI:16521) – a key rate-limiting step in the biosynthesis of cholesterol, steroid hormones, and vitamin D. When cloudy lenses taken from rabbits were treated in vitro with lanosterol, the team found that the clumps of damaged proteins were broken up and the lenses became significantly clearer; use of cholesterol instead of lanosterol was ineffective. In a subsequent in vivo study, they found that administering eye drops containing lanosterol for six weeks to dogs suffering from naturally occurring cataracts resulted in a reduction in the severity of the cataracts and in improved lens clarity.
Although cataract surgery is an effective, generally safe, relatively straightforward and cheap procedure (it is normally performed using a local anaesthetic and takes around 30 to 45 minutes; an allaboutvision.com report found that in 2012, the average cost of straightforward cataract surgery in the US was $3,429 per eye), it nevertheless requires the services of a skilled practitioner. Furthermore, ageing populations around the world are predicted to require a doubling of cataract surgery within 20 years. A drug that would fix a cataract without the need for surgery would therefore represent a major advance.
The background image is a detail from a Creative Commons licensed photograph of a human eye with a blue iris.
Reference(s)
- Goodsell, D.S. (2010) Crystallins RCSB PDB Molecule of the Month, July.
- Global Data on Visual Impairments 2010 (World Health Organisation, Geneva, 2012).
- Cataract surgery (NHS Choices. Health A to Z).
- Zhao, L., Chen, X.-J., Zhu, J., Xi, Y-B., Yang, X., Hu, L.-D., Ouyang, H., Patel, S.H., Jin, X., Lin, D., Wu, F., Flagg, K., Cai, H., Li, G., Cao, G., Lin, Y., Chen, D., Wen, C., Chung, C., Wang, Y., Qiu, A. Yeh, E., Wang, W., Hu, X., Grob, S., Abagyan, R., Su, Z., Tjondro, H.C., Zhao, X.-J., Luo, H., Hou, R., Jefferson, J., Perry, P., Gao, W., Kozak, I., Granet, D., Li, Y., Sun, X., Wang, J., Zhang, L., Liu, Y., Yan, Y.-B. and Zhang, K. (2015) Lanosterol reverses protein aggregation in cataracts Nature, 523 (7562), 607–611.
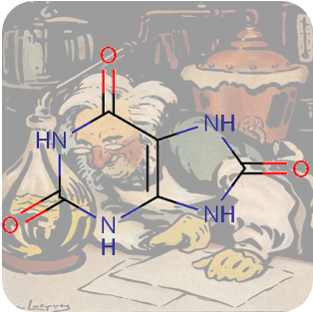
1st August 2015, Uric acid
First discovered by the Swedish chemist Carl Wilhelm Scheele (1742–1786), the heterocyclic metabolite uric acid (UA, CHEBI:27226) is the final product of purine catabolism. In humans, the enzyme xanthine oxidase catalyses the oxidation of the purine, hypoxanthine, to xanthine and subsequently to UA [1]. Most mammals, on the other hand, excrete allantoin and urea as the final purine catabolism products.The levels of UA in blood are a function of the balance between the metabolism of purines and the rate of UA excretion. Typically, the reference range of UA in human blood plasma is 3.4-7.2 mg/dL for men and 2.4-6.1 mg/dL for women. It is estimated that around two thirds of the total body UA is produced endogenously while the remaining one third is derived from dietary purines. Any alterations in normal levels of UA can lead to medical conditions such as gout, type 2 diabetes, Lesch-Nyhan syndrome, cardiovascular disease, and multiple sclerosis [2].
Hyperuricemia, or abnormally high concentration of UA in blood, has been identified as a major worldwide health problem. Clinical studies have associated high levels of UA as predictors of obesity, fatty liver, and diabetes. Research studies have also linked hyperuricemia to the development of metabolic syndrome (disorder of energy utilization and storage), although no strong evidence was reported. This is now changing, however. Recently, several teams of Chinese researchers have established a contributory role of elevated serum UA levels to metabolic syndrome [3]. In a series of experiments on rodents, they found that increased serum levels of UA caused gliosis and hypothalamic inflammation through activation of NF-κB, a protein complex involved in cellular responses to stimuli, eventually leading to systemic metabolic disorders.
The image shows the structure of one of the tautomers of uric acid against a background of a colour photomechanical reproduction of a humerous lithograph by Daniel Thouroude de Losques (1880–1915) depicting the gout specialist Alfred Baring Garrod discovering high concentrations of uric acid in the blood of patients suffering from gout. The file comes from Wellcome Images, a website operated by the Wellcome Trust, a global charitable foundation based in the UK, and is a copyrighted work available under Creative Commons Attribution only licence CC BY 4.0.
Reference(s)
- Gutfreund, H. and Sturtevant, J.M (1959) Steps in the oxidation of xanthine to uric acid catalysed by milk xanthine oxidase. Biochem. J., 73(1), 1–6.
- Borghi, C., Rosei, E.A., Bardin, T., Dawson, J., Dominiczak, A., Kielstein, J.T., Manolis, A.J., Perez-Ruiz, F. and Mancia, G. (2015) Serum uric acid and the risk of cardiovascular and renal disease. J. Hypertens., Jul 5.
- Lu, W., Xu, Y., Shao, X., Gao, F., Li, Y., Hu, J., Zuo, Z., Shao, X., Zhou, L., Zhao, Y. and Cen, X. (2015) Uric acid produces an inflammatory response through activation of NF-κ in the hypothalamus: implications for the pathogenesis of metabolic disorders. Sci. Rep., 5:12144.

1st July 2015, Butyl anthranilate
Insects destroy a very large fraction (thought to be up to 40%) of the global agricultural output and necessitate use of millions of tons of toxic insecticides and insect repellents that are environmentally unfriendly and can be harmful to other species including humans. Finding safe and effective repellents is, therefore, a top priority for agrochemical companies.A significant breakthrough in finding safer alternatives has been made by a research team led by Anandasankar Ray at the University of California, Riverside. They investigated a series of naturally occurring structural analogues of N,N-diethyl-m-toluamide (DEET) [1], a widely used synthetic insect repellent that has been the subject of some safety concerns. One such DEET analogue, butyl anthranilate (BA, CHEBI:86192), has been identified as a potentially safe alternative repellent for the protection of fruit from airborne pests. As well as being relatively expensive, DEET is only meant to be applied to skin and clothing and cannot be sprayed directly on to crops. In a previous study, the team managed to identify the DEET-detecting olfactory receptors that cause the repellence in insect species [2]. Using a computer algorithm, the team were able to identify naturally occurring analogues of DEET with potential to be strong repellents. Several of these were subsequently approved by the US Food and Drug Administration as food additives.
In their current study, the team found that spraying blueberries with a 10% solution of BA, a flavouring and fragrance compound found in several fruits, provided nearly total protection for blueberry samples from a spotted wing Drosophila species, Drosophila suzukii. While most flies are attracted to rotting fruit, D. suzukii feeds specifically on ripening fruit making it a particularly destructive pest. As well as repelling the flies, BA also reduces their desire to lay eggs. Next, Ray wants to test how efficacious BA is in field trials, and if successful, to request approval from the US Environmental Protection Agency. He also intends to explore the potential of extending the approach for protection of humans and livestock from biting insects.
The background image is a detail from a Creative Commons licensed photograph of blueberries.
Reference(s)
- Pham, C.K. and Ray, A. (2015) Conservation of olfactory avoidance in Drosophila species and identification of repellents for Drosophila suzukii. Sci. Rep., 5, doi:srep11527, published online 22 June 2015.
- Kain, P., Boyle, S.M., Tharadra, S.K., Guda, T., Pham C., Dahanukar, A. and Ray, A. (2013) Odour receptors and neurons for DEET and new insect repellents. Nature, 502, 507–512.
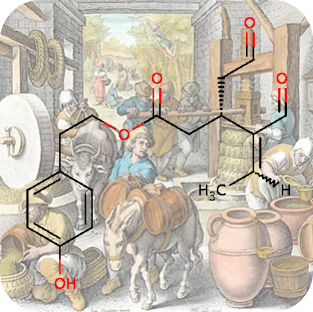
1st June 2015, Oleocanthal
The Mediterranean diet is characterised by a relatively high intake of extra-virgin olive oil (EVOO), fruit, unrefined cereals, and vegetables; moderate amounts of fish, poultry and wine; and relatively low amounts of dairy products, red and processed meats. It has been associated with a number of beneficial health properties, including reduced incidences of cardiovascular disease; age-related cognitive disease; colorectal, prostate, and aerodigestive cancers; and a reduction in overall cancer mortality [1-3].For many years, the health benefits of the diet were attributed to oleic acid, the major component of EVOO. However, when it was shown that the consumption of other seed oils containing similar concentrations of monounsaturated fatty acids does not result in comparable health benefits [4], attention turned to the polyphenolic components of EVOO.
First isolated by Gianfrancesco Montedoro and co-workers at the University of Perugia over twenty years ago [5], a dialdehydic phenol present in EVOO known as (–)-decarboxymethyl ligstroside aglycone was subsequently shown by Paul Andrewes and co-workers at Unilever R&D in Vlaardingen, The Netherlands, to be responsible for the bitterness, pungency and astringency causing the 'throat burn' sensation of many EVOOs [6]. In 2005, Gary Beauchamp and co-workers determined the absolute stereochemistry of the compound and gave it the more convenient name "oleocanthal" (OC; CHEBI:85673) [7].
It has been known for many years that the bitterness of certain compounds correlates with their pharmacological activity [8]. Since oleocanthal caused a similar 'sting in the throat' sensation to the non-steroidal anti-inflammatory drug ibuprofen, Beauchamp's group correctly predicted that, like ibuprofen, OC should have anti-inflammatory properties. They found that not only did OC behave like ibuprofen in inhibiting the action of both cyclooxygenase-1 (COX-1) and COX-2, it was actually significantly more active. Their findings supported the idea that oleocanthal could be a potential factor in a number of the health benefits associated with the Mediterranean diet.
Since COX-2 has been implicated in the pathogenesis of various inflammatory diseases [9,10] and several cancers [11,12] and since OC is a naturally occurring COX-2 inhibitor, it is not surprising that OC has become a compound of significant interest in drug research. Potentially beneficial properties of oleocanthal reported in recent years include the modulation of tau protein fibrilisation [14] and the enhancement of amyloid-β clearance from the brain [15] – both may have important implications for the treatment of neurodegenerative diseases. Most recently, a group led by Prof. David Foster of the Hunter College of the City University of New York has reported on research into the effect of oleocanthal on cultures of human breast, prostate, and pancreatic cancer cell lines. Remarkably, they found that OC rapidly induced cell death in all of the cancer cells studied, whereas with non-cancerous cells, it caused reversible cell cycle arrest – effectively putting the cells to sleep for a while – but did not cause cell death [13].
Oleocanthal is vulnerable to decomposition on exposure to oxygen and light, and is present in olive oil in relatively small amounts (typically between 100 and 160 mg per kg of oil, depending on the time of harvest). Its total synthesis is not currently a viable option as it requires about 10 separate steps, and overall yields are quite low [16,17]. As a consequence, the availability of OC for research has been a problem. Most studies on oleocanthal to date have been in vitro; for in vivo studies, much greater amounts would be required. However, the discovery that significant amounts of OC can be obtained from olive pomace waste [18] and the recent publication of a gentle preparative isolation using high-performance countercurrent chromatography (HPCCC) [19] should help matters.
The background image is a detail from a Creative Commons licensed image depicting the production of olive oil, from an engraving by Philip Galle (1537–1612).
Reference(s)
- Sofi, F., Cesari, F., Abbate, R., Gensini, G.F. and Casini, A. (2008) Adherence to Mediterranean diet and health status: meta-analysis. BMJ [Br. Med. J.], 337(7671), a1344.
- Sofi, F., Abbate, R., Gensini, G.F. and Casini, A. (2010) Accruing evidence on benefits of adherence to the Mediterranean diet on health: an updated systematic review and meta-analysis. Am. J. Clin. Nutr., 92(5), 1189–1196.
- Schwingshackl, L. and Hoffmann, G. (2014) Adherence to Mediterranean diet and risk of cancer: a systematic review and meta-analysis of observational studies. Int. J. Cancer, 135(8), 1884–1897.
- López-Miranda, J., Pérez-Jiménez, F., Ros, E., De Caterina, R., Badimón, L., et al. (2010) Olive oil and health: summary of the II international conference on olive oil and health consensus report, Jaén and Córdoba (Spain) 2008. Nutr., Metab. Cardiovasc. Dis., 20(4), 284–294.
- Montedoro, G., Servili, M., Baldioli, M., Selvaggini, R., Miniati, E. and Macchioni, A. (1993) Simple and hydrolyzable compounds in virgin olive oil. 3. Spectroscopic characterizations of the secoiridoid derivatives. J. Agric. Food Chem., 41(11), 2228–2234.
- Andrewes, P., Busch, J.L.H.C., de Joode, T., Groenewegen, A. and Alexandre, H. (2003) Sensory properties of virgin olive oil polyphenols:âidentification of deacetoxy-ligstroside aglycon as a key contributor to pungency. J. Agric. Food Chem., 51(5), 1415–1420.
- Beauchamp, G.K., Keast, R.S., Morel, D., Lin, J., Pika, J., Han, Q., Lee, C.H., Smith, A.B. and Breslin, P.A. (2005) Ibuprofen-like activity in extra-virgin olive oil. Nature, 437(7055), 45–46.
- Fischer, R., Griffin, F., Archer, R.C., Zinsmeister, S.C. and Jastram, P.S. (1965) Weber ratio in gustatory chemoreception; an indicator of systemic (drug) reactivity. Nature, 207(5001), 1049–1053.
- Parkinson, L. and Keast, R. (2014) Oleocanthal, a phenolic derived from virgin olive oil: a review of the beneficial effects on inflammatory disease. Int. J. Mol. Sci., 15(7), 12323–12334.
- Scotece, M., Conde, J., Abella, V., Lopez, V., Pino, J., Lago, F., Smith, A.B.III, Gómez-Reino, J.J., and Gualillo, O. (2015) New drugs from ancient natural foods. Oleocanthal, the natural occurring spicy compound of olive oil: a brief history. Drug Discovery Today, 20(4), 406–410.
- Ristimäki, A., Sivula, A., Lundin, J., Lundin, M., Salminen, T., Haglund, C., Joensuu, H. and Isola, J. (2002) Prognostic significance of elevated cyclooxygenase-2 expression in breast cancer. Cancer Res., 62(3), 632–635.
- Harris, R.E., Chlebowski, R.T., Jackson, R.D., Frid, D.J., Ascenseo, J.L., Anderson, G., Loar, A., Rodabough, R.J., White, E. and McTiernan, A. (2003) Breast cancer and nonsteroidal anti-inflammatory drugs: prospective results from the Women's Health Initiative. Cancer Res., 63(18), 6096–6101.
- LeGendre, O., Breslin, P.A.S. and Foster, D.A. (2015) (–)-Oleocanthal rapidly and selectively induces cancer cell death via lysosomal membrane permeabilization (LMP). Mol. Cell. Oncol., 2(3462), Published online 23 Jan. 2015.
- Monti, M.C., Margarucci, L., Riccio, R. and Casapullo, A. (2012) Modulation of tau protein fibrillization by oleocanthal. J. Nat. Prod., 75(9), 1584–1588.
- Abuznait, A.H., Qosa, H., Busnena, B.A., El Sayed, K.A. and Kaddoumi, A. (2013) Olive-oil-derived oleocanthal enhances β-amyloid clearance as a potential neuroprotective mechanism against Alzheimer's disease: in vitro and in vivo studies. ACS Chem. Neurosci., 4(6), 973–982.
- Smith, A.B.III, Sperry, J.B. and Han, Q. (2007) Syntheses of (–)-oleocanthal, a natural NSAID found in extra virgin olive oil, the (–)-deacetoxy-oleuropein aglycone, and related analogues. J. Org. Chem., 72(18), 6891–6900.
- Valli, M., Peviani, E.G., Porta, A., D'Alfonso, A., Zanoni, G. and Vidari, G. (2013) A concise and efficient total synthesis of oleocanthal. Eur. J. Org. Chem, (20), 4332–4336.
- Cicerale, S., Conlan, X.A., Barnett, N.W. and Keast, R.S. (2011) The concentration of oleocanthal in olive oil waste. Nat. Prod. Res., 25(5), 542–548.
- Adhami, H.R., Zehl, M., Dangl, C., Dorfmeister, D., Stadler, M., Urban, E., Hewitson, P., Ignatova, S. and Krenn, L. (2015) Preparative isolation of oleocanthal, tyrosol, and hydroxytyrosol from olive oil by HPCCC. Food Chem., 170, 154–159.

1st May 2015, Neonicotinoid Insectide
In our Entity of the Month article for May 2012, we highlighted the possible link between the use of neonicotinoid insecticides (CHEBI:25540) and the rapid decline in the bee population, known as colony collapse disorder. Two separate reports showed that exposure to sub-lethal amounts of neonicotinoids resulted in much lower production of queens [1] as well as interference with the bees' ability to navigate and communicate [2], which are vital for the survival of colonies. As a result, in 2013 the European Union (EU) introduced a temporary ban on the use of the three neonicotinoids, clothianidin (CLO), imidacloprid (IMD) and thiamethoxam (TMX), on flowering crops, while further scientific and technical evidence was gathered. The EU's restrictions have been disputed by agrichemical companies and national governments who have questioned the validity of the studies. Now, two new studies have provided a further twist in the saga of neonicotinoid pesticides and their disputed effects on bee health.A key argument of those who originally opposed the ban was that bees in their natural environment would simply choose not to feed on the nectar from neonicotinoid-treated plants. To test this assumption, a research team led by Professor Geraldine Wright at Newcastle University, UK, used neuron recording techniques which showed that neither honeybees (Apis mellifera) nor bumblebees (Bombus terrestris ) were able to identify neonicotinoids by their bitter taste and were not repelled by them. The bees were then offered a choice of sugar solutions, with some contaminated by neonicotinoids at levels similar to those found in the nectar of treated crop plants. Surprisingly, both honeybees and bumblebees showed a preference for some of the pesticide-tainted solutions [3]. The effect was more pronounced with bumblebees and was observed with IMO and TMX, but not CLO. This work shows that bees cannot control their exposure to neonicotinoids in food and implies that treating flowering crops with IMD and TMX presents a significant danger to foraging bees.
A separate field study carried out by a research team led by Maj Rundlöf at Lund University in Sweden focussed on how neonicotinoids influence wild bees in in real-life agricultural environments [4]. They used Elado, a commercial insecticide that contains CLO, in field tests with rapeseed and found at the treated sites there were reductions in wild bee density, solitary bee nesting activity and bumblebee reproduction. While these effects were observed in wild bees, no significant impact was seen in honeybees, the subject of many previous studies. The findings indicate that the contribution of pesticides to the global decline of wild bees may have been underestimated. Also, the lack of a significant response in honeybee colonies suggests that reported pesticide effects on honeybees cannot always be extrapolated to wild bees. Hence, EU officials will have plenty of reading material to consider before the current ban expires.
The image is a Creative Commons licensed picture of A close-up of a bee in the grounds of Langdon Court near Wembury.
Reference(s)
- Stokstad, E. (2012). Field research on bees raises concern about low-dose pesticides. Science, 335, 1155.
- Whitehorn, P.R., O'Connor, S., Wackers, F.L. and Goulson D. (2012). Neonicotinoid pesticide reduces bumble bee colony growth and queen production. Science, 336, 351–352.
- Kessler, S.C., Tiedeken, E.J., Simcock, K.L., Derveau, S., Mitchell, J., Softley, S., Stout, J.C. and Wright, G.A. (2015). Bees prefer foods containing neonicotinoid pesticides. Nature, doi:10.1038/nature14414, published online 22 April 2015.
- Rundlöf, M., Andersson, G.K.S., Bommarco, R., Fries, I., Hederström, V., Herbertsson, L., Jonsson, O., Klatt, B.K., Pedersen, T.R., Yourstone, J. and Smith, H.G. (2015). Seed coating with a neonicotinoid insecticide negatively affects wild bees. Nature, doi:10.1038/nature14420, published online 22 April 2015.
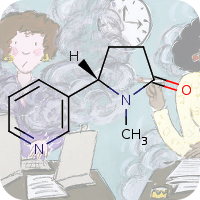
1st April 2015, (–)-Cotinine
Second-hand smoke (SHS), also known as passive or environmental tobacco smoke (ETS), is the involuntary inhalation of the smoke produced by cigarettes and other tobacco products in homes, cars, offices and public places. It is a complex mixture of approximately 4000 toxic chemicals including carcinogens such as 4-aminobiphenyl, 2-naphthylamine, benzene and a variety of polycyclic aromatic hydrocarbons.Classified as a Group A carcinogen by the United States Environmental Protection Agency (EPA) over 20 years ago [1], exposure to SHS has been shown to cause carcinogenic, mutagenic and cardiovascular effects in various species of animals [2]. Inhalation of tobacco smoke is particularly dangerous for children, resulting in increased incidence of respiratory infections, bacterial meningitis, asthma and cot death [3]. It is estimated that every year in the UK alone, passive smoking causes over 165,000 new cases of disease in children and kills over 12,000 people from lung cancer and various other respiratory diseases [4].
A number of active analytical techniques have been developed to assess non-smokers' exposure to tobacco, including estimation of levels of environmental markers such as carbon monoxide, nicotine, particulate matter, etc. in air samples, the prediction of smoke concentrations by mathematical representations or exposure models, or determination of biomarkers in vivo [5]. Owing to its high sensitivity and specificity, the measurement of cotinine (CHEBI:68641) in human biofluids has been found to be the most useful biomarker in assessing exposure to SHS [6]. Commonly found in the leaves of the tobacco plant Nicotiana tabacum, cotinine is the peripheral oxidative metabolite of nicotine which is rapidly metabolised in the liver and converted into cotinine by enzymes such as cytochrome P450 2A6 [7].
In a recent study, a team of researchers led by Aderonke Akinkugbe at The University of North Carolina at Chapel Hill, USA, have found an association between serum cotinine levels and periodontitis in US non-smokers [8]. In an analysis involving 3,255 non-smoking volunteers, it was concluded that the non-smokers exposed to ETS were more susceptible to moderate/severe periodontitis than the unexposed. Their findings have been presented in a paper titled "Environmental Tobacco Smoke is Associated with Periodontitis in US Non-Smokers" at the 93rd General Session and Exhibition of the International Association for Dental Research.
The background image shows a detail from a 1993 cartoon titled 'I mind very much if you smoke', depicting two women in a working situation, with one of the women smoking. The image is in the public domain and can be freely reused. It was release by the National Cancer Institute, an agency part of the National Institutes of Health, with the ID of 8049.
Reference(s)
- EPA designates passive smoking a "Class A" or known human carcinogen. EPA press release 7 January 1993.
- Joya, X., Manzano, C., Álvarez, A.T., Mercadal, M., Torres, F., Salat-Batlle, J. and Garcia-Algar, O. (2014) Transgenerational exposure to environmental tobacco smoke. Int. J. Environ. Res. Public Health, 11(7), 7261–7274.
- Treyster, Z. and Gitterman, B. (2011). Second hand smoke exposure in children: environmental factors, physiological effects, and interventions within pediatrics. Rev. Environ. Health, 26(3), 187–195.
- Passive smoking. (Cancer Research UK).
- Jaakkola, M.S. and Jaakkola, J.J. (1997) Assessment of exposure to environmental tobacco smoke. Eur. Respir. J., 10(10), 2384–2397.
- Murphy, S.E., Wickham, K.M., Lindgren, B.R., Spector, L.G. and Joseph, A. (2013) Cotinine and trans 3'-hydroxycotinine in dried blood spots as biomarkers of tobacco exposure and nicotine metabolism. J. Exposure Sci. Environ. Epidemiol., 23(5), 513–518.
- Dwoskin, L.P., Teng, L., Buxton, S.T. and Crooks, P.A. (1999) (S)-(–)-Cotinine, the major brain metabolite of nicotine, stimulates nicotinic receptors to evoke [3H]dopamine release from rat striatal slices in a calcium-dependent manner. J. Pharmacol. Exp. Ther, 288(3), 905–911.
- Environmental tobacco smoke is associated with periodontitis in US non-smokers. ScienceDaily 13 March 2015.
1st March 2015, Ellagic acid
First discovered by the French pharmacist Henri Braconnot in 1831 [1], the polyphenol antioxidant ellagic acid (EA, CHEBI:4775) is found in many fruits and nuts, including blackberries, cranberries, grapes, pomegranates, raspberries, strawberries, pecans, and walnuts. It has also been found in many species of oak [2], the Eurasian watermilfoil (Myriophyllum spicatum) [3], and the Japanese medicinal mushroom Phellinus linteus [4].Active research programmes into the biological properties of ellagic acid have been ongoing for over half a century. In the 1960s, most research was related to its effect on blood clotting [5], while from the 1970s onwards, numerous studies on EA and cancer were published. As a result, EA has been marketed as a dietary supplement with benefits claimed against a variety of medical problems, including cancer and heart disease. However, there is a vast difference between compounds found to inhibit cancer in cell cultures or animal studies and an effective treatment for the disease in real patients, and in 2008, the US Food and Drug Administration (FDA) included ellagic acid in a list of "187 Fake Cancer Cures Consumers Should Avoid".
In the last decade, two new uses for ellagic acid have been described. One is as an alternative to compounds such as hydroquinone or kojic acid in skin lightening creams [6]. The other could be of far greater significance. In 2012, a group led by Associate Professor Liwei Gu of the Food Science and Human Nutrition Department of the Institute of Food and Agricultural Sciences at the University of Florida fed some mice on a diet containing 10% fat, and others on a 60% fat diet. Some of each group were also fed an extract of muscadine grapes. (The mice particularly like the high-fat diet and over-consume it, so are regarded as a good model for people with a sedentary lifestyle who eat too much junk food and don't get enough exercise). The mice on the high fat diet developed fatty livers and symptoms of diabetes, but those that were also given the grape extract accumulated much less fat in their livers and had lower blood sugars [7]. In a recent follow-up paper, Gu and co-workers have shown that ellagic acid is a particularly potent component of the muscadine grape extract. By exposing human liver and fat cells grown in the laboratory to EA, the group found that new fat cell formation and fatty acid biosynthesis in adipose tissue was reduced, while in the liver the synthesis of triglycerides and fatty acids was reduced and fatty acid oxidation was increased [8].
With one in four adults now classed as obese, the UK is the fattest country in Europe [9]. As a result, obesity and its associated diseases (coronary heart disease, type 2 diabetes, liver and gall bladder disease) make up the biggest public health crisis facing the country. The possible development of a dietary strategy for reducing the harmful accumulation of fat in the liver suggested by the work of Gu and co-workers would be very welcome news.
The background image is a a Creative Commons licensed photograph of two white mice – one normal, the other extremely fat.
Reference(s)
- Grasser, G. (1922). Synthetic Tannins, their synthesis, industrial production, and application. The Technical Press, London. p20.
- Mämmelä, P., Savolainen, H., Lindroos, L., Kangas, J. and Vartiainen, T. (2000) Analysis of oak tannins by liquid chromatography-electrospray ionisation mass spectrometry. J. Chromatogr. A, 891(1), 75–83.
- Nakai, S., Inoue, Y., Hosomi, M. and Murakami, A. (2000) Myriophyllum spicatum-released allelopathic polyphenols inhibiting growth of blue-green algae Microcystis aeruginosa. Water Res., 34(11), 3026–3022.
- Lee, Y.S., Kang, Y.-H., Jung, J.-Y., Lee, S., Ohuchi, K., Shin, K.H., Kang, I.-J., Park, J.H.Y., Shin, H.-K. and Lim, S.S. (2008) Protein glycation inhibitors from the fruiting body of Phellinus linteus. Biol. Pharm. Bull., 31(10), 1968–1972.
- Bock, P.E., Srinivasan, K.R. and Shore, J.D. (1981) Activation of intrinsic blood coagulation by ellagic acid: insoluble ellagic acid-metal ion complexes are the activating species. Biochemistry, 20(25), 7258–7266.
- Dahl, A., Yatskayer, M., Raab, S. and Oresajo, C. (2013) Tolerance and efficacy of a product containing ellagic and salicylic acids in reducing hyperpigmentation and dark spots in comparison with 4% hydroquinone. J. Drugs Dermatol., 12(1), 52–58.
- Gourineni, V., Shay, N.F., Chung, S., Sandhu, A.K. and Gu, L. (2012) Muscadine grape (Vitis rotundifolia) and wine phytochemicals prevented obesity-associated metabolic complications in C57BL/6J mice. J. Agric. Food Chem., 60(31), 7674–7681.
- Okla, M., Kang, I., Kim, D.M., Gourineni, V., Shay, N., Gu, L. and Chung, S. (2015) Ellagic acid modulates lipid accumulation in primary human adipocytes and human hepatoma Huh7 cells via discrete mechanisms. J. Nutr. Biochem, 26(1), 82–90.
- The State of Food and Agriculture 2013. (PDF, 2.44Mb) United Nations Food and Agricultural Organization. p 79.

1st February 2015, Cytisine
The alkaloid cytisine (CHEBI:4055) has been used as an anti-smoking aid in Eastern European countries for several decades. Cytisine is obtained from the seeds of the common laburnum (golden rain) tree, Laburnum anagyroides, and is a partial agonist of nicotinic acetylcholine receptors. It was discovered in 1818, first isolated in 1865, and its actions were documented as "qualitatively indistinguishable from that of nicotine" in 1912 [1].Cytisine was used as a cheap tobacco substitute by German and Russian soldiers during the Second World War, and was brought to market in 1964 as an anti-smoking aid under the brand name Tabex, now produced by the Bulgarian drug company Sopharma. Studies on its effects have been published since the 1960s, but none of the early trials conformed to Western regulatory standards and hence did not lead to wider adoption of the drug. Moreover, the pre-clinical studies necessary to define the optimal dosage to be used in humans were never conducted. Large scale studies using current methodology were thus missing for the drug to be registered according to modern US and European standards. However, it is noteworthy that varenicline, a structural analogue of cytisine, was introduced as a smoking cessation drug by Pfizer in 2006.
The first modern study of cytisine was carried out in 2011 by a team led by Robert West at University College London and concluded that it helped smokers more than treble their chances of quitting compared to a placebo [2]. Now researchers in New Zealand have carried out a follow-up trial comparing cytisine with standard nicotine replacement therapy (NRT). The team, led by Natalie Walker at the University of Auckland National Institute for Health Innovation, examined 1,310 smokers who were trying to quit. Half of the participants were given cytisine as tablets, which were to be taken in diminishing doses every day for 25 days, while the other half were given regular NRT in the form of patches, gums, or lozenges, for two months. The researchers found that people taking cytisine were actually far more successful in stopping smoking than those on NRT; after six months, 143 of the 655 cytisine patients were still abstaining from smoking while only 100 in the NRT group of 655 had remained tobacco-free [3]. Although some side effects, including nausea, vomiting and sleep disturbance were observed in subjects using cytisine, these were never significant. In addition, cytisine is considerably less expensive than other available anti-smoking aids and hence will have potential for use in poorer regions of the world where people are unable to afford NRT treatment.
The background image is of an 1879 lithograph of an illustration by the American comic artist Thomas Worth (1834–1917) depicting a smoker exhaling cigar smoke which forms the words "try one". The file comes from Wellcome Images, a website operated by the Wellcome Trust, a global charitable foundation based in the UK, and is a copyrighted work available under Creative Commons Attribution only licence CC BY 4.0.
Reference(s)
- Dale, H.H. and Laidlaw, P.P. (1912). The physiological action of cytisine, the active alkaloid of laburnum (Cytisus laburum). J. Pharmacol. Exp. Ther., 3, 205–221.
- West, R., Zatonski, W., Cedzynska, M., Lewandowska, D., Pazik, J., Aveyard, P. and Stapleton, J. (2011). Placebo-controlled trial of cytisine for smoking cessation. N. Eng. J. Med.,365, 1193–1200.
- Walker, N., Howe, C., Glover, M., McRobbie, H., Barnes, J., Nosa, V., Parag, V., Bassett, B. and Bullen, C. (2014). Cytisine versus nicotine for smoking cessation. N. Eng. J. Med.,371, 2353–2362.
1st January 2015, Testosterone
Testosterone (CHEBI:17347) was first isolated from bull testes in 1935 by Ernst Lanquer and a team of scientists at Organon International, based in Oss, in the south of the Netherlands [1]. The results of their experiments were published as "On crystalline Male Hormone from Testicles (testosterone)", coining the name for the newly isolated hormone (from stems of testicle and sterol, and the suffix of ketone). Biologically synthesised in the Leydig cells of testes and adrenal glands, the effects of testosterone can be classified as virilising (involved in biological development of sexually differentiating characteristics) and anabolic (growth of muscle mass and strength, bone density and maturation).Biosociological and psychological models as well as studies on animal models suggest that testosterone concentration is often related to aggressiveness and dominance in a variety of species including primates. Sociologist and engineer Allan Mazur, Professor of Public Affairs in the Maxwell School of Syracuse University, New York, proposed that the levels of testosterone and dominance are directly related – dominant individuals show a higher level of testosterone than those exhibiting submissiveness [2]. A pre-competitive surge in testosterone levels during sporting events and high testosterone levels in winners relative to that in the losers' suggest "winner effect" of testosterone in humans [3]. However, a new research study by David Edwards, Professor of Psychology at Emory University and graduate student Kathleen Casto has raised doubts on this claim [4].
When Edwards and Casto analysed the saliva samples of participants of an inter-collegiate cross country running competition, they found levels of testosterone in athletes surged during the race irrespective of the their finishing time. The researchers therefore proposed that the increased levels of hormones during competitions may be indicators of a psychological strength rather than being related to results of a competition.
The background image is a detail from a Creative Commons licensed photo of the cross country race on 28 April 1940 "Criterium des veterans". Henri Smets (at right on the photo) was the winner.
Reference(s)
- Freeman, E.R., Bloom, D.A. and McGuire, E.J. (2001) A brief history of testosterone. J. Urol., 165(2), 371–373.
- Mazur, A. (2013). Biosocial model of status in face-to-face primate groups. Procedia Soc. Behav. Sci., 84, 53–56.
- Booth, A., Shelley, G., Mazur, A., Tharp, G. and Kittok, R. (1989) Testosterone, and winning and losing in human competition. Horm. Behav., 23(4), 556–571.
- Casto, K.V., Elliott, C.M. and Edwards, D.A. (2014) Intercollegiate cross country competition: Effects of warm-up and racing on salivary levels of cortisol and testosterone. Int. J. Exerc. Sci. , 7(4), 318–328.
2014
1st December 2014, Ergometrine
The fungus Claviceps purpurea is probably best known as the cause of ergot, a fungal disease of rye and other cereals in which black elongated fruiting bodies grow in the ears of the cereal, and which was once a dreaded pest. When infected grain is milled, the fungal fruiting bodies, known as sclerotia, are ground up into a dark red powder. While its presence in light-coloured flour is readily apparent, it is very easily missed in dark rye flour. The alkaloids produced by the fungus are known as ergot alkaloids and are very diverse in terms of both structure and biological activity, and outbreaks of poisoning from their ingestion through the consumption of rye bread made from ergot-infected grain was once common, especially in wet seasons when the rye is particularly prone to infection.Symptoms of ergot poisoning are both convulsive (painful seizures, diarrhoea, nausea and vomiting, followed by mania or psychosis) and gangrenous (vasoconstriction causing excruciating pain in fingers, toes, and even whole limbs, loss of peripheral sensation, skin peeling from affected tissues and their eventual death and loss). Cattle were also affected, while pregnant sows littered prematurely – an observation particularly relevant to this month's Entity. An early, vivid description comes from the year 857 in the Annales Xantenses: "a Great Plague of swollen blisters consumed the people by a loathsome rot, so that their limbs were loosened and fell off before death". During the Middle Ages, ergot poisoning acquired the common name 'St. Anthony's Fire', after the monks of the Hospital Brothers of St. Anthony, an order founded in the late 11th Century with the purpose of caring for those suffering from ergot poisoning. The relationship between the disease and ergot of rye was recognised by the 16th Century, but epidemics occurred throughout Europe until well into the 19th Century, when careful monitoring of rye and procedures for removing and preventing ergot were developed. The danger of crop damage and intoxication remains, however, and occasional outbreaks of ergot poisoning still occur in poorer regions of the world.
Despite the risk of poisoning, controlled doses of ergot were used for centuries to induce abortions and to reduce maternal bleeding after childbirth. In 1917, Arthur Stoll (1887–1971), a young Swiss chemist (and student of the 1915 Nobel Prize winner Richard Willstätter), was given the task of setting up a new pharmaceutical department by the chemical company Sandoz. He proposed isolating the oxytocic component of Claviceps sclerotia. However, isolation of the compound by solvent extraction proved to be very challenging: the methods available for characterising compounds were restricted to characteristic colour reactions, elemental analysis, melting points, and optical rotations, as none of the spectroscopic techniques in use today existed at that time. Keller's reagent (1896), and in later work Van Urk's reagent (1929) [1], which give specific colours for indoles substituted at position 4, were particularly useful, but elemental analysis was the most reliable measurement. Melting points had to be treated with caution as they may vary by several degrees due to the presence of trace amounts of solvents or other impurities, while the use of optical rotation measurements actually hindered progress – it is now known that all ergot alkaloids are laevorotatory, but that during extraction and isolation they can become dextrorotatory as a result of light, temperature, or pH. By 1918, Stoll had isolated the principal alkaloid produced by Claviceps sclerotia and the first ergot alkaloid ever to be isolated in a pure state. Although Stoll's compound, ergotamine, was active, it was apparent that it was not the compound he had been looking for. Although it was used as an oxytocic drug for some time, the results were poor.
Stoll stopped working on the problem until 1932, when he heard that a Scottish researcher, John Chassar Moir (1900–1977), a reader in obstetrics and gynaecology at University College Hospital in London, had found that the oxytocic activity of Claviceps remained in the aqueous phase after the main alkaloids had been removed by solvent extraction [2]. Moir teamed up with the English biochemist Harold Ward Dudley (1887–1935) to work on the compound's extraction, which the pair achieved in 1935; their compound was only the second ergot alkaloid isolated. Sadly, Dudley died on the day that the first paper describing the work was published [3]. In a reference to its action on the endometrium, Moir and Dudley named the new alkaloid ergometrine (CHEBI:4822) [3,4]. Three other groups reported the compound's isolation in the same year: Morris Kharasch (1895-1957) and R.R. Legault from the University of Chicago proposed the name ergotocine [5], Marvin R. Thompson from Johns Hopkins University in Baltimore pointed out that he had reported the isolation of the same substance in his Ph.D. thesis a year earlier and used the name ergostetrine [6], while Stoll suggested ergobasine, in reference to the alkaloid's basic character [7]. After exchanging products, the researchers agreed that they had all isolated the same compound, and that differences in the melting point and optical rotation measurements were due to differences in purity [8]. Unfortunately, they couldn't agree on which name to use. The American researchers opted for ergonovine, but the English and Swiss groups didn't agree, with the result that the names ergonovine, ergobasine, and ergometrine occur in the literature even today, although use of ergometrine prevails.
From 1935 onwards, preparations of ergometrine have been administered to pregnant women (commonly as the maleate salt) in conjunction with oxytocin to induce uterine contractions in the third stage of labour (delivery of the placenta). Its action is more prolonged than that of oxytocin, and the contractions induced are particularly intense. It is also used to prevent or control postpartum (or postabortal) haemorrhage – by maintaining uterine contraction, blood vessels in the uterine wall are compressed, so reducing blood flow. In so doing, it has saved countless lives. Since 1935, the maternal death rate in England and Wales has fallen from around 400 per 100,000 live births [9] to less than 10 in 100,000 [10]. While many factors have contributed to the decline, the use of ergometrine has undoubtedly been a major player – worldwide, haemorrhage is the main cause of maternal deaths [11].
All drugs have side effects, and egometrine is no exception. These are generally minor but unpleasant (headache, abdominal pain, diarrhoea, nausea, tinnitus, palpitations) but doctors must also be alert for hypertension and cardiac arrhythmias. Recently, however, a paper by Amy Brown and Sue Jordan of the College of Human and Health Sciences at Swansea University has highlighted another consequence [12]. In a study of 288 mothers, no significant association was found between the choice of infant feeding mode (breast or formula milk) and the use of prophylactic uterotonics (oxytocin or ergometrine) at birth. Just 2 weeks later, however, the mothers who had been treated with prophylactic uterotonics were significantly less likely to be breastfeeding. The same was true four weeks later. When mothers who had abandoned breast feeding were asked why, those in the 'uterotonics' group were more likely to cite pain and difficulty than those in the control group. The authors suggest that mothers who receive uterotonics may need additional support to establish breastfeeding.
The background image is a detail from a of a 4 fluid ounce (ca. 120 ml) bottle of ergot extract produced by Parke Davis & Company Ltd.,1891–1950. The file comes from Wellcome Images, a website operated by the Wellcome Trust, a global charitable foundation based in the UK, and is a copyrighted work available under Creative Commons Attribution only licence CC BY 4.0.
Reference(s)
- van Urk, H.W. (1929) Een nieuwe gevoelige reactie op de moederkoornalkaloiden ergotamine, ergotoxine en ergotinine en de toepassing voor het onderzoek en de colorimetrische bepaling in moederkoornpreparaten. Pharm. Weekbl., 66(22), 473–481.
- Moir, C., and Dale, H.H. (1932) The action of ergot preparations on the puerperal uterus. A clinical investigation with special reference to an active constituent of ergot as yet unidentified. Brit. Med. J., 1(June 18), 1119–1122.
- Dudley, H.W. and Moir, C. (1935) The substance responsible for the traditional clinical effect of ergot. Brit. Med. J., 1(March 16), 520–523.
- Dudley, H.W. and Moir, J.C. (1935) New active principle of ergot. Science, 81(7 June), 559–560.
- Kharash, M.S. and Legault, R.R. (1935) Ergotocin: the active principle of ergot responsible for the oral effectiveness of some ergot preparations on human uteri. J. Am. Chem. Soc., 57(5), 956–957.
- Thompson, M.R. (1935) The new active principle of ergot. Science, 81(2113), 636–639.
- Stoll, A. (1935) The new ergot alkaloid. Science, 82(2131), 415–417.
- Kharasch, M.S., King, H., Stoll, A. and Thompson, M.R. (1936) New ergot alkaloid. Nature, 137(3462), 403–405.
- Barker, M.E. (1935) Annual report of the Medical Officer of Health and School Medical Officer for the year 1935. County Borough of East Ham. p 14.
- London 'has high rate of childbirth deaths'. (NHS Choices Health News). April 30, 2012.
- Lalonde, A, Daviss, B.A., Acosta, A. and Herschderfer, K. (2006) Int. J. Gynecol. Obstet., 94(3), 243–253.
- Brown, A. and Jordan, S. (2014) Active management of the third stage of labor may reduce breastfeeding due to pain and physical complications. Breastfeed. Med., 9(10), published online October 27 2014.
3rd November 2014, Tramadol
In our Entity of the Month article for October 2013, we highlighted the surprising discovery by Michel De Waard and co-workers at Joseph Fourier University in Grenoble that the opioid painkiller tramadol (CHEBI:9648), thought to be synthetic, was actually being produced in nature by the African plant Nauclea latifolia (commonly known as the 'pincushion tree') [1]. Although several pharmaceuticals have been discovered in plants after being synthesised in the laboratory, this was the first time a drug had been found in clinically significant (reported as 0.4%) concentrations. However, a new research project has cast doubt on this claim.De Waard's results attracted the attention of the analytical chemist Michael Spiteller at Dortmund University of Technology in Germany. Together with an international team of collaborators, he reported that the appearance of tramadol in the pincushion tree could concievably be a result of contamination caused by human activity [2]. Firstly, he contacted De Waard for a sample he could analyse independently. His African colleagues collected samples from Cameroon's semi-arid northern region as well as its southern rain forest, completely different locations from De Waard's. Conducting their own analysis, Spiteller's group confirmed that De Waard's sample contained 0.4% tramadol. In contrast, none of their own samples were found to contain more than 0.00015% tramadol. Furthermore, samples from southern Cameroon contained no detectable tramadol at all. Team members based in Cameroon conversed with local farmers and learned that tramadol is readily available at low cost from local markets or street sellers in northern Cameroon and that farmers in the north regularly feed tramadol to cattle so they can work for longer periods at high temperatures. This practice was not reported in the south of the country. Spiteller's team proposes that cattle dosed with tramadol would excrete in areas under pincushion trees, and the synthetic tramadol be subsequently taken up by the plant roots. Mass spectrometry of the northern pincushion tree roots, as well as water and soil samples from surrounding areas, confirmed the presence of tramadol and its mammalian metabolites. In addition, they found trace quantities of both tramadol and its metabolites in the roots of other unrelated plants in the same areas.
The team leader of the original study, however, is standing by his results. De Waard states that his samples were taken from a national park where livestock is prohibited and also suggests that baboons and other mammals in the national park would eat the fruits from the pincushion trees, which may explain the presence of metabolites in the soil. He also suggests that differing plant ages, symbiotic fungi or microbes, and Cameroon's varied climate could explain the discrepancy in the teams' results [3]. The next step is to carry out plant feeding studies with isotope- or radiolabelled compounds. If labelled tramadol fed to cattle ends up in plant roots, then that would provide strong evidence for tramadol to be a contaminant. But if the plant can use labelled precursor compounds to synthesise tramadol, that would suggest tramadol is naturally produced.
The background image is a Creative Commons licensed picture showing the fruit and flower of Nauclea latifolia.
Reference(s)
- Boumendjel, A., Taiwe, G.S., Bum, E.N., Chabrol, T., Beney, C., Sinniger, V., Haudecoeur, R., Marcourt,L., Challal, S., Queiroz, E.F., Souard, F., Le Borgne, M., Lomberget, T., Depaulis, A., Lavaud, C., Robins,R., Wolfender, J.-L., Bonaz, B. and De Waard., M. (2013) Occurrence of the synthetic analgesic tramadol in an African medicinal plant. Angew. Chem. Int. Ed., 52, 11780–11784.
- Kusari, S., Tatsimo, S.J.N., Zühlke, S., Kouam, S.F. and Spiteller, M. (2014) Tramadol–a true natural product? Angew. Chem. Int. Ed., doi:10.1002/anie.201406639, published online 12 September 2014.
- Drahl, C. (2014) Tramadol's newfound natural product status in doubt. Chem. Eng. News, 92, 34–35.
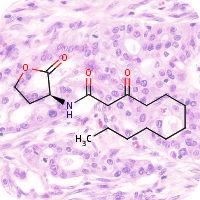
6th October 2014, N-(3-Oxododecanoyl)-L-homoserine lactone
Deaths from pancreatic cancer rank fifth amongst cancer-related mortalities in the UK. According to cancer statistics from the world's largest independent cancer research charity, Cancer Research UK, approximately 8,700 people (4,300 men and 4,400 women) in the UK died from pancreatic cancer in 2012, equivalent to nearly 24 victims every day [1]. The disease is more common in the elderly with an increased incidence among patients with diabetes, chronic pancreatitis, and obesity [2]. It has been estimated that about 29% of cases of pancreatic cancers in the UK are linked to tobacco smoking [3]. The disease has poor prognosis and statistically, the overall five-year survival rate of the cancer victims is less than 5% [4]. Insights into the tumour pathogenesis show that a mature pancreatic cell harbours an average of 63 genetic alterations which makes the disease genetically complex and heterogeneous [5]. In the early stages, the symptoms of the cancer are quite common and non-specific (e.g. pain in the back or abdomen); symptoms sufficiently specific to lead doctors to suspect pancreatic cancer tend to occur only when the cancer is already at an advanced stage. The cancer is generally resistant to chemotherapy [6] while the surgical removal of a part or the entire pancreas known as pancreatectomy, usually suggested for prolonged survival and cure, is associated with undue perioperative risk and significant morbidity [7]. The challenge therefore remains to find therapies or novel molecules that would ensure better outcomes.Quorum sensing (QS) is a type of inter-cellular communication especially within a bacterial community that is used to optimise the metabolic and behavioural activities of a colony of bacteria for life in close proximity [8]. This form of communication is mediated by a group of small extracellular signalling molecules called autoinducers (AIs). The concentration of AIs varies as a function of the bacterial cell density; when sufficient bacteria are present, the concentration of AIs reaches a threshold level that allows the bacteria to sense a critical cell mass and in turn activate or repress target genes. This differential gene regulation enables bacteria to express characteristic behaviour only while growing in social communities. Both Gram-positive and Gram-negative bacteria use quorum sensing systems to regulate a multitude of traits such as biofilm formation [9], production of virulence factors [10], motility [11], and antibiotic resistance.
Research studies have further revealed an inter-kingdom signalling whereby the bacterial QS molecules may modify or stimulate the behaviour of the host eukaryotic cells [12]. Pseudomonas aeruginosa, an opportunistic pathogen, produces several such QS molecules including members of the N-acyl homoserine lactone family that induce virulence gene expression (i.e. the expression of genes that contribute to the organism's ability to cause disease). In particular, the lipophilic molecule N-(3-oxododecanoyl)-L-homoserine lactone (CHEBI:44534) (O-DDHSL) has been found to interact with a range of mammalian cells such as respiratory epithelial cells, T cells and antigen-presenting cells (APCs) [13]. It has also been shown that O-DDHSL inhibits proliferation and induces apoptosis (programmed cell death) in human breast carcinoma cell lines [14]. Based on these reports, a team of scientists led by Professor Senthil Kumar,of the Comparative Oncology and Epigenetics Laboratory at the University of Missouri College of Veterinary Medicine proposed that such signalling molecules could be used to 'tell' cancer cells to stop spreading [15]. The study analysed the migration, viability and colonisation of pancreatic carcinoma cells in vitro and the effect of alteration of genes involved in these processes following O-DDHSL treatment. Results showed a decrease in cell viability from apoptosis and diminished colony formation, along with an inhibition of migration of the cancer cells, indicating that the QS chemical could be used as an effective biomolecule for targeting pancreatic cancer. The researchers now intend to find ways to use this molecule outside laboratory settings to validate the efficacy of the therapy under real life conditions.
We have chosen N-(3-oxododecanoyl)-L-homoserine lactone as our entity of month in support of Macmillan Cancer Support, one of the largest cancer support charities in the UK, who have recently organised the World's Biggest Coffee Morning, an annual fundraising event spread over numerous locations in the UK, including our Hinxton Campus. Readers may also be interested in the forthcoming 'Purple Lights for Hope' pancreatic cancer awareness initiative taking place across the UK on November 1st 2014.
The background image is a detail from a Creative Commons licensed histopathogic depiction of pancreatic adenocarcinoma arising in the pancreas head region.
Reference(s)
- Cancer statistics key facts. Pancreatic cancer, Cancer Research UK, September 2014.
- Lowenfels, A.B. and Maisonneuve, P. (2006) Epidemiology and risk factors for pancreatic cancer. Best Pract. Res., Clin. Gastroenterol., 20(2), 197–209.
- Pancreatic cancer risk factors. (Cancer Research UK).
- England and Wales survival (2011-2012) summary. (Cancer Research UK).
- Jones, S., Zhang, X., Parsons, D.W., Lin, J.C., Leary, R.J., Angenendt, P., Mankoo, P., et al. (2008) Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science, 321(5897), 1801–1806.
- Qanungo, S., Uys, J.D., Manevich, Y., Distler, A.M., Shaner, B., Hill, E.G., Mieyal, J.J., et al. (2014) N-acetyl-L-cysteine sensitizes pancreatic cancers to gemcitabine by targeting the NFκB pathway. Biomed. Pharmacother., doi: 10.1016/j.biopha.2014.08.007, published online 28 August 2014.
- Bliss, L.A., Witkowski, E.R., Yang, C.J. and Tseng, J.F. (2014) Outcomes in operative management of pancreatic cancer. J. Surg. Oncol., 110(5), 592–598.
- Sifri, C.D. (2008) Healthcare epidemiology: quorum sensing: bacteria talk sense. Clin. Infect. Dis., 47(8), 1070–1076.
- Galante, J., Ho, A.C., Tingey, S. and Charalambous, B.M. (2014) Quorum sensing and biofilms in the pathogen, Streptococcus Pneumoniae. Curr. Pharm. Des., published online 5 September 2014.
- Bala, A., Chhibber, S. and Harjai, K. (2014) Pseudomonas quinolone signalling system: A component of quorum sensing cascade is a crucial player in the acute urinary tract infection caused by Pseudomonas aeruginosa. Int. J. Med. Microbiol., doi:10.1016/j.ijmm.2014.08.013, published online 1 September 2014.
- Yang, Q. and Defoirdt, T. (2014) Quorum sensing positively regulates flagellar motility in pathogenic Vibrio harveyi. Environ. Microbiol., doi:10.1111/1462-2920.12420, published online 15 February 2014.
- Hughes, D.T. and Sperandio, V. (2008) Inter-kingdom signalling: communication between bacteria and their hosts. Nat. Rev. Microbiol., 6, 111–120.
- Ritchie, A.J., Whittall, C., Lazenby, J.J., Chhabra, S.R., Pritchard, D.I. and Cooley, M.A. (2007) The immunomodulatory Pseudomonas aeruginosa signalling molecule N-(3-oxododecanoyl)-L-homoserine lactone enters mammalian cells in an unregulated fashion. Immunol. Cell Bio.l, 85 (8), 596–602.
- Li, L., Hooi, D., Chhabra, S.R., Pritchard, D. and Shaw, P.E. (2004) Bacterial N-acylhomoserine lactone-induced apoptosis in breast carcinoma cells correlated with down-modulation of STAT3. Oncogene, 23 (28), 4894–4902.
- Kumar, A.S., Bryan, J.N. and Kumar, S.R. (2014) Bacterial quorum sensing molecule N-3-oxo-dodecanoyl-L-homoserine lactone causes direct cytotoxicity and reduced cell motility in human pancreatic carcinoma cells. PLoS One, 9 (29), e106480.
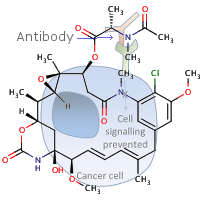
1st September 2014, Maytansine
First isolated in 1972 from the African plant Maytenus ovatus, later named Maytenus serrata, by Professor S. Morris Kupchan and co-workers at the University of Virginia using bioassay-guided methods, the 19-membered macrolactam maytansine (CHEBI:6701) was found to combine broad spectrum antitumour activity with extraordinarily high potency [1]. However, it took a further five years before a more efficient isolation procedure was developed using M. buchananii and sufficient quantities of maytansine became available to enable clinical trials to proceed. In the meantime, with funding particularly from the US National Cancer Institute, a number of closely related compounds known as 'maytansinoids' were isolated from members of the bittersweet (Celastraceae), buckthorn (Rhamnaceae), and spurge (Euphorbiaceae) plant families. Unfortunately, early hopes that maytansine or maytansinoids could be readily developed into a clinically useful anticancer drug were dashed by disappointing Phase II clinical trials, probably a result of the dose-limiting cytotoxicity of the maytansinoids in humans.What was required was a means to selectively deliver the cytotoxic maytansinoid to the tumour cells, leaving healthy cells alone. This is the idea behind antibody-drug conjugates: a cytotoxic drug is joined to an antibody that specifically targets a certain marker in the tumour – such as a protein which, ideally, is only found in tumour cells. The antibodies bind to the tumour cells and in so doing trigger the tumour cell to absorb the antibody and its attached cytotoxin. Once inside the tumour cell, the cytotoxin is released, causing the death of the cell. Since healthy cells are largely spared the effects of the toxin, the side effects commonly associated with traditional chemotherapy such as hair loss, nausea, and fatigue should also be reduced.
In one approach to finding a suitable maytansinoid-antibody conjugate, Genentech, a subsidiary of Roche since 2009, chose the monoclonal antibody trastuzumab (brand names Herclon and Herceptin), which binds to the HER2/neu receptor, an important biomarker for around 20-30% of breast cancer patients [2]. To complete the conjugate, Genentech attached the maytansinoid mertansine to the antibody. The resulting conjugate, trastuzumab emtansine (also known as ado-trastuzumab emtansine; brand name Kadcyla) attacks breast cancer in two ways. The trastuzumab stops the cancer cells from overexpressing the HER2 protein, stopping them from growing and spreading, while the attached cytotoxic mertansine fragment kills the cancerous cells. Trials found that, although trastuzumab emtansine was not a cure, it could help control the disease: the median overall survival time of women with HER2-positive breast cancer who were already resistant to trastuzumab alone was almost 6 months longer using trastuzumab emtansine than the previous best treatment [3] and the US Food and Drug Administration approved trastuzumab emtansine for the treatment of HER2-positive advanced breast cancer (i.e. cancer that has spread to other parts of the body, cannot be surgically removed and has stopped responding to initial treatment) in February 2013 [4].
The improved outcome for HER2-positive breast cancer patients comes at a price, however. Excluding VAT and administration costs, a course of treatment of trastuzumab emtansine for an average period of 14.5 months for a typical patient (70.1 kg; weekly dose 3.6 mg/kg) would cost over £90,000 - many times that of current treatments [5]. In the UK, an estimated 8,300 women and 60 men are diagnosed with HER2-positive breast cancer each year [5], so the cost to the National Health Service of the trastuzumab emtansine treatment would be substantial. The question is, could these costs be justified by the benefits of the improved outcomes? The National Institute for Health and Care Excellence (NICE) is charged with the task of determining whether new treatments, procedures, diagnostic agents, etc., represent value for money. In general, NICE will not recommend a treatment as cost-effective unless it costs less than £30,000 per quality-adjusted life year (QALY). The QALY value of trastuzumab emtansine was estimated to be around £166,000. Even though Roche proposed a discount to the full list price of the drug, it was not sufficient to affect the outcome. Last month, the NICE Appraisal Committee submitted their final determination to the Institute, recommending rejection of the drug [6]. It follows a similar decision from Ireland's equivalent pricing body, the National Centre for Pharmacoeconomics (NCPE Ireland), in June [7]. While Roche have complained about the way in which cancer drugs are appraised, pointing out that NICE have rejected the last eight breast cancer drugs in a row, it seems unlikely that the funding formula is going to be overhauled anytime soon.
The background image is a detail from a Creative Commons licensed depiction of an antibody, such as trastuzumab, blocking a receptor, such as the HER2/neu receptor, on the surface of a cancer cell, so preventing cell signalling.
Reference(s)
- Kupchan, S.M., Komoda, Y., Court, W.A., Thomas, G.J., Smith, R.M., Karim, A., Glimore, C.J., Hartiwanger, R.C.,and Bryan, R.F. (1972) Maytansine, a novel antileukemic ansa macrolide from Maytenus ovatus. J. Am. Chem. Soc., 94(4), 1354–1356.
- Mitri, Z., Constantine, T. and O'Regan, R. (2012) The HER2 receptor in breast cancer: pathophysiology, clinical use, and new advances in therapy. Chemother. Res. Pract., 743193.
- Verma, S., Miles, D., Gianni, L., Krop, I.E., Welslau, M., Baselga, J., Pegram, M., Oh, D.-Y., Diéras, V., Guardino, E., Fang, L., Lu, M.W., Olsen, S. and Blackwell, K. (2012) Trastuzumab emtansine for HER2-positive advanced breast cancer. New Engl. J. Med., 367(19), 1783–1791.
- FDA approves new treatment for late-stage breast cancer (FDA press release), February 22, 2013.
- Kadcyla: NICE disappointed by manufacturer's decision (NICE press release), August 7, 2014.
- Final appraisal determination - trastuzumab emtansine for treating HER2-positive, unresectable locally advanced or metastatic breast cancer after treatment with trastuzumab and a taxane. National Institute for Health and Care Excellence, August 2014.
- Trastuzumab emtansine summary National Centre for Pharmacoeconomics, June 30, 2014.
4th August 2014, Psilocybin
Psilocybin (CHEBI:8614) is the active ingredient in more than 200 species of mushrooms, collectively known as psilocybin mushrooms, psychedelic mushrooms or magic mushrooms, that have long been widely used for their hallucinogenic properties. A prodrug, it is rapidly converted by the body to the active metabolite psilocin which has mind-altering effects that are similar (in some aspects) to those observed with other hallucinogenics such as lysergic acid diethylamide (LSD) and mescaline. Although these compounds are non-addictive, prolonged usage and overdose can lead to brain damage and reactions such as anxiety, paranoia and delusions. However, scientists have been unsure as to exactly what happens to the brain when under the influence of psychedelic drugs.Now, a team of researchers led by Enzo Tagliazucchi at Goethe University Frankfurt have found that psilocybin induces changes in the brain that are similar to those that occur during dreams [1]. The researchers injected 15 volunteers with liquid psilocybin while they were lying in a functional magnetic resonance imaging (fMRI) scanner and compared the scans with images taken when the same people were given a placebo. This revealed that while under influence of the drug, there was increased activity in the hippocampus and anterior cingulate cortex. These areas of the brain are involved in emotions and the formation of memories, and were among the first to evolve. The team also observed decreased activity in the more recently evolved regions of the brain associated with self-control and higher thinking, such as the thalamus, posterior cingulate and medial prefrontal cortex. This activation pattern is similar to that observed when someone is dreaming. The researchers now intend to utilise their findings to explore the potential use of hallucinogens for treatment of depression.
The background image is a Creative Commons licensed picture of Psilocybe weilii.
Reference(s)
- Tagliazucchi, E., Carhart-Harris, R., Leech, R., Nutt, D. and Chialvo, D.R. (2014). Enhanced repertoire of brain dynamical states during the psychedelic experience. Hum. Brain Mapp., DOI: 10.1002/hbm.22562, published online 2 July 2014.

7th July 2014, Tetrodotoxin
Tetrodotoxin (CHEBI:9506), commonly abbreviated to TTX, is a low molecular weight neurotoxin characteristically distributed in both terrestrial and marine animal kingdoms [1]. It derives its name from Tetraodontiformes (tetras-four and odontos-tooth), an order that includes pufferfish, porcupinefish, ocean sunfish and several other such species in which the toxin is found. TTX is generally present in the gonads, liver, intestines and skin of these species, the amount fluctuating according to the species, region and seasonal variations [2]. Although TTX is found in these fish and other animals (e.g. gastropods, newts, blue-ringed octopuses etc.), studies have revealed that it is actually synthesised by endo-symbiotic bacteria (e.g. Pseudoalteromonas tetraodonis, certain species of Pseudomonas and Vibrio, etc.) that naturally inhabit the gut of the animal. It has been proposed that the animal initially acquires the TTX-producing bacteria via the food web [3].Tetrodotoxin poisoning has been primarily attributed to the consumption of puffer fish, a culinary delicacy in Japan and other regions along the coast of Asia that is locally known as fugu [4]. The toxin is thermally stable and cannot be destroyed by cooking or storage under freezing temperatures. On ingestion, it blocks the voltage-gated sodium channel, thus inhibiting the production and propagation of action potentials, mainly in skeletal muscles, neurons and nerve fibres [5]. Clinical manifestations of tetrodotoxication (puffer fish poisoning) include shortness of breath, numbness, tingling, light-headedness, paralysis, irregular heartbeat and eventual death. TTX is several times more potent than cyanide: the lethal dose in humans is just 2-3 mg. When taken in near-lethal doses, it leaves the person in a state of impending death; it is colloquially known as "zombie powder", particularly in the voodoo and Haitian cultures. British explorer, Captain James Cook was the first person to record TTX poisoning in 1774 after an incident of ingestion of puffer fish liver near Polynesian islands. Amusingly, Ian Fleming, best known for his James Bond series of spy novels, also appeared to have a special attraction for TTX as it features in two of his novels – From Russia, with Love and Dr. No – as a chemical used for an assassination attempt on the MI6 agent [6].
In the 1890s, D. Takahashi, the first professor of pharmacology at the University of Tokyo, worked towards the pharmacological characterisation and identification of the toxic principle from puffer fish. However, it was not until 1909 that the Japanese scientist Dr. Yoshizumi Tahara isolated the purified toxin from the ovaries of the pufferfish and coined the name “tetrodotoxin” [7]. The chemical structure of TTX wasn't finally solved until 1964, when three independent studies led by the great American synthetic organic chemist Robert Burns Woodward [8] at Harvard, Prof. Toshio Goto of the Chemical Institute of Nagoya University [9] and Prof. Kyosuke Tsuda of the Institute of Applied Microbiology (now the Institute of Molecular and Cellular Biosciences) at the University of Tokyo [10] each published the correct structure.
Recently, a research group led by Amber Stokes from California State University, Bakersfield, California, has established the occurrence of the neurotoxin TTX for the first time in terrestrial invertebrates. The group found that two terrestrial flatworm species, Bipalium adventitium and Bipalium kewense, use TTX in predation to immobilise their prey [11]. However, little knowledge is currently available regarding the production or acquisition of the toxin in the tissues of the organisms.
The background image is a Creative Commons licensed picture of a puffer fish, Arothron hispidus, at the Big Island of Hawaii.
Reference(s)
- Yasumoto, T., Nagai, H., Yasumura, D., Michishita, T., Endo, A., Yotsu, M. and Kotaki, Y. (1986) Interspecies distribution and possible origin of tetrodotoxin. Ann. N. Y. Acad. Sci., 479, 44–51.
- Deeds, J. (2012) Tetrodotoxin, in Bad Bug Book. Foodborne Pathogenic Microorganisms and Natural Toxins. Second Edition., 211–217. Food and Drug Administration
- Bane, V., Lehane, M., Dikshit, M., O’Riordan, A. and Furey, A. (2014) Tetrodotoxin: chemistry, toxicity, source, distribution and detection. Toxin, 6(2), 693–755.
- Kheifets, J., Rozhavsky, B., Girsh Solomonovich, Z., Marianna, R. and Soroksky, A. (2012) Severe tetrodotoxin poisoning after consumption of Lagocephalus sceleratus (Pufferfish, Fugu) fished in Mediterranean sea, treated with cholinesterase Inhibitor. Case Rep. Crit. Care, 2012, 782507.
- Moczydlowski, E.G. (2013) The molecular mystique of tetrodotoxin. Toxicon, 63, 165–183.
- Moore, J.W. and Narahashi, T. (1967) Tetrodotoxin's highly selective blockage of an ionic channel. Fed. Proc., 26(6), 1655–1663.
- Suehiro, M. (1994) Historical review on chemical and medical studies of globefish toxin before World War II. Yakushigaku Zasshi, 29(3), 428–434.
- Woodward, R.B. (1964) The structure of tetrodotoxin. Pure Appl. Chem., 9(1), 49–74.
- Goto, T., Kishi, Y., Takahashi, S. and Hirata, Y. (1965) Tetrodotoxin. Tetrahedron, 21(8), 2059–2088.
- Tsuda, K., Ikuma, S., Kawamura, M., Tachikawa, R. and Sakai, K. (1964) Tetrodotoxin. VII. On the structure of tetrodotoxin and its derivatives. Chem. Pharm. Bull., 12(11), 1357–1374.
- Stokes A.N., Ducey, P.K., Neuman-Lee, L., Hanifin, C.T., French, S.S., Pfrender, M.E., Brodie, E.D.3rd and Brodie, E.D.Jr. (2014) Confirmation and distribution of tetrodotoxin for the first time in terrestrial invertebrates: two terrestrial flatworm species (Bipalium adventitium and Bipalium kewense). PLoS One, 9(6), e100718.

2nd June 2014, Vitamin E
At the University of California, Berkeley in 1922, the pioneering histologist and medical researcher Katharine Scott Bishop (1889–1975) and the anatomist and endocrinologist Herbert McLean Evans (1882–1971) found that rats did not reproduce if they were fed a semi-synthetic purified diet in which lard was the sole source of fat, but that this fertility defect was corrected by a 'substance X' present in lipid extracts of various grains [1]. The essential dietary component required to prevent such fertility defects is known today as vitamin E (CHEBI:33234), a term which covers four tocopherols (named from the Greek tokos, birth, and pherein, to bear or carry, so meaning 'to carry a pregnancy') and four tocotrienols. Both groups are based on a chroman-6-ol skeleton which is substituted by a hydrocarbon chain at position 2, and are designated as α-, β-, γ-, and δ- depending on the number and position of methyl groups attached to the aromatic ring. The most active compound is (+)-α-tocopherol. First isolated in a pure form in 1935 by Gladys Anderson Emerson (1903–1984), its structure was elucidated by Erhard Fernholz (1909–1940) at Princeton in 1938 [2]; the first synthesis was published later that year by the Swiss organic chemist and Nobel Prize winner Paul Karrer (1889–1971) and his team [3,4].Found particularly in olive and sunflower oils, α-tocopherol is the main source of vitamin E in the European diet and in dietary supplements. However, a higher intake of rapeseed (canola), soya bean (soybean) and maize (corn) oils means that in the American diet, the most common form is γ-tocopherol. The two forms differ by only one methyl group, but that is sufficient to confer distinct biological properties on each form. Thus while both forms correct the birth defect studied by Bishop and Evans, and both have a relatively similar capacity to scavenge reactive oxygen species [5,6], γ-tocopherol also reacts with reactive nitrogen species, whereas α-tocopherol does not [7].
At the Division of Allergy and Immunology of the Feinberg School of Medicine at Northwestern University in Chicago, a group led by Associate Professor Joan Cook-Mills has previously shown that, in mice, α-tocopherol supplementation reduces the symptoms of eosinophilic lung inflammation and airway hyperresponsiveness, whereas γ-tocopherol supplementation worsens the symptoms [8,9]. The group later found a mechanism to explain the difference – while both α- and γ-tocopherol bind to protein kinase Cα, α-tocopherol inhibits its action whereas γ-tocopherol increases its action [10].
Following a major study involving over 4,500 participants, Cook-Mills and her team have recently found that the different properties of α- and γ- tocopherol also influence human health. While consumption of α-tocopherol was associated with improved lung function, the study found that consumption of γ-tocopherol was associated with an increased incidence of lung inflammation and, possibly, asthma [11]. It is perhaps no coincidence that the rise in rates of asthma in the US over the last 40 years coincides with a switch in US diets from lard and butter to soya bean, rapeseed and maize oils (which had been thought to be healthier for the heart). The average blood plasma level of γ-tocopherol in the US is more than four times that in southern European and Scandinavian countries where sunflower and olive oils are consumed, and where asthma rates are significantly lower.
The image is a Creative Commons licensed picture of Katharine Scott Bishop, who co-discovered vitamin E with Herbert Evans.
Reference(s)
- Evans, H.M. and Bishop, K.S. (1922) On the existence of a hitherto unrecognized dietary factor essential for reproduction. Science (Washington, DC, U. S.), 56(1458), 650–651.
- Fernholz, E. (1938) On the constitution of α-tocopherol. J. Am. Chem. Soc., 60(3), 700–705.
- Karrer, P., Fritzsche, H., Ringier, B.H. and Salomon, H. (1938) α-Tocopherol. Helv. Chim. Acta, 21(1), 520–525.
- Karrer, P., Fritzsche, H., Ringier, B.H. and Salomon, H. (1938) Synthese des α-Tocopherols. Helv. Chim. Acta, 21(1), 820–825.
- Yoshida, Y., Saito, Y., Jones, L.S. and Shigeri, Y. (2007) Chemical reactivities and physical effects in comparison between tocopherols and tocotrienols: physiological significance and prospects as antioxidants. J. Biosci. Bioeng., 104(6), 439–445.
- Nishio, K., Horie, M., Akazawa, Y., Sichiri, M., Iwahashi, H., Hagihara, Y., Yoshida, Y. and Niki, E. (2013) Attenuation of lipopolysaccharide (LPS)-induced cytotoxicity by tocopherols and tocotrienols. Redox Biol., 1(1), 97–103.
- Patel, A., Liebner, F., Netscher, T., Mereiter, K. and Rosenau, T. (2007) Vitamin E chemistry. Nitration of non-α-tocopherols: products and mechanistic considerations. J. Org. Chem., 72(17), 6504–6512.
- Berdnikovs, S., Abdala-Valencia, H., McCary, C., Somand, M., Cole, R., Garcia, A., Bryce, P. and Cook-Mills, J.M. (2009) Isoforms of vitamin E have opposing immunoregulatory functions during inflammation by regulating leukocyte recruitment. J. Immunol., 182(7), 4395–4405.
- Cook-Mills, J.M. and McCary, C.A. (2010) Isoforms of vitamin E differentially regulate inflammation. Endocr., Metab. Immune Disord.: Drug Targets, 10(4), 348–366.
- McCary, C.A., Yoon, Y., Panagabko, C., Cho, W., Atkinson, J. and Cook-Mills, J.M. (2012) Vitamin E isoforms directly bind PKCα and differentially regulate activation of PKCα. Biochem. J., 441(1), 189–198.
- Marchese, M.E., Kumar, R., Colangelo, L.A., Avila, P.C., Jacobs, D.R.Jr., Gross, M., Sood, A., Liu, K. and Cook-Mills, J.M. (2014) The vitamin E isoforms α-tocopherol and γ-tocopherol have opposite associations with spirometric parameters: the CARDIA study. Respir. Res., 15, 31.
6th May 2014, 5α-androst-16-en-3-one
The role of serendipity has been a common occurrence throughout the history of scientific innovation and discovery. Examples include Alexander Fleming's accidental discovery of penicillin in 1928 and the invention of the microwave oven by Percy Spencer in 1945. Recently, John McGlone of Texas Tech University in Lubbock, who studies animal behaviour, has discovered that the pig pheromone 5α-androst-16-en-3-one (commonly known as androstenone, CHEBI:37894) can be employed to control the behaviour of unruly or over-exuberant dogs. The discovery was made when McGlone sprayed his own pet dog, which was barking excessively, with the pheromone and was intrigued when the barking suddenly stopped.The findings were studied further by the research team [1]. Dogs were sprayed using an aerosol containing androstenone, while simultaneously exposing them to a loud noise that would normally frighten or excite them. Compared to a spray of alcohol and noise alone, the androstenone was found to be much more effective at calming the dogs. In a second study, the dogs' heart rates were monitored and were found to be unaltered by treatment with androstenone, indicating that they were not scared or stressed. McGlone has subsequently worked with pet care company Sergeant's to develop the androstenone spray, which is now commercially available as a training tool.
Androstenone is found in boar saliva and helps induce sows to mate. In dogs, it works through the olfactory system, acting as an interomone; that is, a substance that is produced by one species but has an effect on the behaviour of a different species. As yet, it is unclear whether the effect on dogs is actually anything to do with androstenone being a pig pheromone.
The background image is a Creative Commons licensed picture showing Buster, a two-year old Lhasa Apso.
Reference(s)
- McGlone, J.J., Thompson, W.G. and Guay, K.A. (2014) Case Study: The pig pheromone androstenone, acting as an interomone, stops dogs from barking. Prof. Anim. Sci., 30,105–108.

7th April 2014, Dimethyl sulfide
Plant defence against herbivory defines a cascade of morphological, biochemical, and molecular alterations in plants to improve their survival and reproduction by reducing herbivore preference and performance [1]. Plant resistance against herbivores can be either constitutive (expressed irrespective of the external stimuli) or induced in response to an attack by the predator. Inducible plant defence is a paradigm of phenotypic plasticity that elicits physiological or behavioural changes in the invader [2]. In turn, the impact of plant resistance on the herbivores can be either direct or indirect. The resistance factors for direct plant defence include morphological features such as thorns, spines, and prickles, trichomes [3], wax-coated cell walls, as well as secretions like gummosis or sap that trap insect predators [4]. Plants also produce toxic chemicals such as insect repellents or feeding deterrents that can adversely affect the growth and development of herbivores [5]. Indirect methods of defence include tritrophic interactions, whereby plants protect themselves by attracting natural enemies of the herbivores such as parasitoids and predators. This is mediated via a release of volatile cues, commonly known as herbivore-induced plant volatiles (HIPVs), by the plants when attacked [6].HIPVs primarily comprise of terpenoids, fatty acid derivatives and phenylpropanoids and can be emitted either from injured plant tissues, or systematically from uninjured tissues. Also known as infochemicals, the HIPVs communicate between the infested plant and the natural carnivorous enemies of the attacking herbivores as well as neighbouring plants and different parts of the damaged plants (inter- and intra-plant signalling respectively) [7].
According to a recent paper published on the study of mutualistic interactions in the Southern Ocean by Matthew Savoca and Gabrielle A. Nevitt from Department of Neurobiology, Physiology, and Behavior at University of California, Davis, dimethyl sulfide (DMS, CHEBI:17437), widely studied in the context of regulation of the global climate (the ‘CLAW’ hypothesis – the acronym is made up from the first letter of the surnames of its proponents), mediates tritrophic interactions between phytoplankton (a group of photosynthesising microorganisms including diatoms, dinoflagellates, cyanobacteria and algae; the primary producers) and procellariiformes (an order of seabirds which include albatrosses, petrels and shearwaters; top predators) [8]. DMS, produced mainly from the algal metabolite dimethylsulfoniopropionate (DMSP) during lysis of the algal cell triggered by grazing crustaceans such as krill (primary consumers), is known to attract some species of procellariiform seabirds. These carnivorous species in turn selectively forage on krill. Additionally, the defecation by marine top predators serves as a potential source of recycled iron for the phytoplankton. The challenge now is to understand the response of such marine ecosystems to the possible extinction of marine top predators – of the 21 albatross species on the International Union for Conservation of Nature IUCN Red List, 19 are threatened and the other two are "near threatened" – and the subsequent loss of their contribution to the trace-nutrient recycling [9].
The background image is a Creative Commons licensed picture showing a Northern krill (Meganyctiphanes norvegica).
Reference(s)
- Cory, J.S. and Hoover, K. (2006) Plant-mediated effects in insect-pathogen interactions. Trends Ecol. Evol., 21(5), 278–286.
- War, A.R., Paulraj, M.G., Ahmad, T., Buhroo, A.A., Hussain, B., Ignacimuthu, S. and Sharma, H.C. (2012) Mechanisms of plant defense against insect herbivores. Plant Signaling Behav., 7(10), 1306–1320.
- Riddick, E.W. and Simmons, A.M. (2014) Do plant trichomes cause more harm than good to predatory insects? Pest Manage. Sci., doi: 10.1002/ps.3772, published online 1 March 2014.
- Skrzypek, E., Miyamoto, K., Saniewski, M. and Ueda, J. (2005) Identification of jasmonic acid and its methyl ester as gum-inducing factors in tulips. J. Plant Res., 118(1), 27–30.
- Ibanez, S., Gallet, C. and Després, L. (2012) Plant insecticidal toxins in ecological networks. Toxins, 4(4), 228–243.
- War, A.R., Sharma, H.C., Paulraj, M.G., War, M.Y. and Ignacimuthu, S. (2011) Herbivore induced plant volatiles: their role in plant defense for pest management. Plant Signaling Behav., 6(12), 1973–1978.
- Arimura, G., Matsui, K. and Takabayashi, J. (2009) Chemical and molecular ecology of herbivore-induced plant volatiles: proximate factors and their ultimate functions. Plant Cell Physiol., 50(5), 911–923.
- Savoca, M.S. and Nevitt, G.A. (2014) Evidence that dimethyl sulfide facilitates a tritrophic mutualism between marine primary producers and top predators. Proc. Natl. Acad. Sci. U. S. A., doi: 10.1073/pnas.1317120111, published online 18 March 2014.
- Fitzgerald, K.T. (2013) Longline fishing (how what you don't know can hurt you). Top. Companion Anim. Med., 28(4), 151–162.
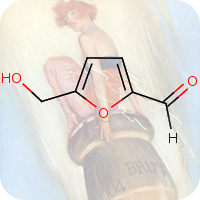
3rd March 2014, 5-Hydroxymethylfurfural
5-Hydroxymethylfurfural (CHEBI:412516), commonly abbreviated to 5-HMF, was one of the compounds that was studied by Louis-Camille Maillard (1878–1936) during his investigations into the browning reaction that now bears his name [1] (although the first coherent scheme to explain the various products did not appear for another 40 years [2]). The Maillard reaction occurs during the heating of substances containing reducing_sugars, such as D-glucopyranose, and free amino groups (of amino acids or, in proteins, mainly the ε-amino group of L-lysine, but also the α-amino groups of terminal amino acids). It is of particular importance in the food industry, as its products play an important part in the aroma, taste and colour of foods prepared by baking, roasting or toasting.Although 5-HMF is virtually absent from fresh food, it is naturally generated as a Maillard reaction product during the drying or cooking of sugar-containing food via acid-catalysed thermal dehydration of D-fructofuranose, sucrose, and, to a lesser degree, D-glucopyranose. It is estimated that in a Western diet, intakes of 5-HMF generally range between 4 and 30 mg per person per day, although individual levels can vary widely – an intake of up to 350 mg can result from beverages made from dried plums [3]. However, it is the presence of 5-HMF in sparkling wines, rather than foods, together with the potential benefits of this to the wine producers, that has recently made news.
Sparkling wines are produced by treating a blend of base wines with sugar and yeasts and allowing the resulting mixture to ferment, thereby introducing enough carbon dioxide into the wine to make it effervescent. The fermentation can be carried out either within sealed bottles (the traditional or Champenoise method, used for Champagne, Cava, Crémant and Spumante), or in steel tanks, with the product being bottled under pressure in a continuous process (the Charmat method, used for Asti, Prosecco and Sekt). Whichever method is used, and however good the resulting product, the producers cannot easily determine how long its shelf life will be. It is known that environmental factors such as temperature are very important, but measuring the degree to which a bottle of wine has aged is problematic. Wineries currently use the 'browning' of their wine as the basis of their tests. Wine browning during its ageing and storage is a result of a non-enzymatic process of oxidation and polymerisation of the phenolic compounds present in the wine: determining the wine's absorbance of light at a wavelength of 420 nm therefore provides a simple and rapid method for measuring the amount of browning that has occurred in a sample of wine. While the technique is successful with white wines, however, the grapes used in the production of many sparkling wines tend to give pale- or straw-yellow products, with the consequence that the absorbance test is not very sensitive [4].
Now a group led by Montserrat Riu-Aumatell of the Institute for Research on Nutrition and Food Safety (INSA-UB) at the University of Barcelona has found a solution to the problem. They analysed samples of six sparkling wines kept at refrigerator (4 °C), cellar (16 °C) and room (20 °C) temperature for absorbance at 420 nm, phenolic components, and 5-HMF at regular intervals over a period of two years. They found that the amount of 5-HMF gave the clearest indication as to the freshness of the wines, showing a linear increase over time at each temperature for all of the wines tested. They also noted that refrigerating sparkling wines almost entirely prevents browning. To make their results of more practical use to the wineries, the group also developed a mathematical model that allows producers to predict the shelf life of sparkling wines as a function of storage time and temperature [5].
The background image is a Creative Commons licensed picture of 1915 English magazine illustration of a lady riding a champagne cork.
Reference(s)
- Maillard, L.-C. (1912) Action des acides aminés sur les sucres. Formation des melanoidins par voie methodique. Compt. Rend., 154, 66–68
- Hodge, J.E. (1953) Dehydrated foods. Chemistry of browning reactions in model systems. J. Agric. Food Chem., 1(15), 928–943.
- Abraham, K., Gûrtler, R., Berg, K., Heinemeyer, G., Lampen, A. and Appel, K.E. (2011), Toxicology and risk assessment of 5-hydroxymethylfurfural in food. Mol. Nutr. Food Res., 55(5), 667–678.
- Serra-Cayuela, A., Aguilera-Curiel, M.A., Riu-Aumatell, M., Buxaderas, S. and López-Tamames, E. (2013) Browning during biological aging and commercial storage of Cava sparkling wine and the use of 5-HMF as a quality marker.Food Res. Int., 53(1), 226–231.
- Serra-Cayuela, A., Jourdes, M., Riu-Aumatell, M., Buxaderas, S., Teissedre, P.-L. and López-Tamames, E. (2014) Kinetics of browning, phenolics, and 5-hydroxymethylfurfural in commercial sparkling wines. J. Agric. Food Chem., 62(5), 1159–1166.

3rd February 2014, ω–3 fatty acid
The flesh of oily fish such as mackerel and salmon, plus the livers of white fish such as cod, are the main sources of ω–3 fatty acids (CHEBI:25681), which are known to be essential components of the human diet. The most important ones are icosapentaenoic acid (also known as eicosapentaenoic acid and most commonly abbreviated to EPA) and docosahexaenoic acid (DHA), which are thought to provide a range of benefits to human health, including lowering of cholesterol levels and reducing the risk of coronary heart disease, cancer, depression and attention-deficit hyperactivity disorder. Fish do not actually produce ω–3 fatty acids themselves. In the ocean, they accumulate them by eating smaller fish that have in turn eaten algae, the only marine organisms that produce significant quantities of EPA and DHA. Farmed fish, that have no access to these algae, are instead fed fishmeal enriched with fish oil containing EPA/DHA. However overfishing, climate change and ocean acidification have diminished global fish stocks, resulting in supplies of high quality fish oil becoming limited. Hence alternative and sustainable sources of ω–3 fatty acids are needed to relieve some of the pressure on the oceans.One potential source has been unearthed by Professor Johnathan Napier and colleagues at Rothamsted Research in Harpenden, UK, The team have genetically modified a biofuel plant called Camelina sativa, the seeds of which are known to be rich in α-linolenic acid, a key intermediate in the biosynthesis of EPA and DHA [1]. They took seven genes from the algae that produce these fatty acids and inserted them into the genome of C. sativa. The seeds of the modified plant yielded oil that, when purified, contained around 12% EPA and 14% DHA, the same proportions as in fish oil [1]. The team anticipate that the plant oil could be available commercially within 10 years. It could then help replace the fish oil used in capsules or fed to farmed fish.
In a separate study, Rolf Müller and colleagues at Saarland University have found that certain species of myxobacteria (also known as slime bacteria after the slime they produce to aid their movement) possess the genes necessary to synthesise certain ω–3 fatty acids by employing multi-enzyme systems known as polyunsaturated fatty acid (PUFA) synthases [2]. Müller's team has so far discovered two distinct species: Sorangium cellulosum can make linoleic acid (a key precursor for EPA and DHA) while the recently discovered genus Aetherobacter has been found to produce prolific amounts of EPA and DHA. Although both strains grow slowly and are difficult to handle, the team report that the genes can be transferred to and expressed by Myxococcus xanthus, a fast growing model strain of myxobacteria [2]. The next challenge will be to exploit or engineer this natural biosynthetic pathway to develop an economically feasible process for production of EPA and DHA.
This month's image is a Creative Commons licensed picture showing a migrating school of mackerel.
Reference(s)
- Ruiz-Lopez, N. Haslam, R.P., Napier, J.A. and Sayanova, O. (2014) Successful high-level accumulation of fish oil omega-3 long-chain polyunsaturated fatty acids in a transgenic oilseed crop. Plant. J., 77,198–208.
- Gemperlein, K., Rachid, S., Ronald O. Garcia, R.O., Wenzel, S.C. and Müller, R. (2014) Polyunsaturated fatty acid biosynthesis in myxobacteria: Different PUFA synthases and their product diversity.DOI:10.1039/C3SC53163E, published online 2 January 2014.

4th January 2014, Tannin
Tannins (CHEBI:26848) are the polyphenolic compounds of relatively high molecular weight that have the ability to form complexes with proteins and various other organic compounds [1]. Generally associated with the ripening of fruits [2] and organoleptic properties of drinks such as wine [3] and tea, tannins are the ubiquitous plant-based substances responsible for the astringent taste found in many foods and beverages.The term "tannin" originates from the ancient practice of using extracts from trees to convert animal hide to leather, a process commonly known as tanning. This technique exploits the inherent property of the tannins to bind with various macromolecules. When applied to animal skin, tannins bind to the collagen proteins making the hide imputrescible (resistant to decay) and flexible for end use.
Tannins are the most abundant secondary metabolites found principally in the bark, leaves and immature fruits of species throughout the plant kingdom, where they play a role defending the plant against herbivores by deterrence or toxicity. Structurally, they can be classified as hydrolysable tannins (HTs) and condensed tannins (CTs, also known as proanthocyanidins). The HTs generally consist of a carbohydrate core partially or totally esterified with phenolic groups such as in gallic acid (in gallotannins) or ellagic acid (in ellagitannins). CTs, however, are polymers of flavonoid units joined together by non-hydrolysable carbon-carbon bonds.
Research on tannin-herbivore interactions has played a significant role in the field of ecology due to the possible influence of tannins on the feeding behaviour of many invertebrate and vertebrate herbivores [4]. In fact, high levels of tannins in tree leaves are commonly believed to increase the resistance of trees to insects. This has been attributed to the interference of the tannins with the digestion in the insect herbivore by inhibition of enzymes, metabolic activities of symbiotic microorganisms (rumen microbes) that contribute to digestion, or by reducing the digestibility of ingested nutrients. According to a recent theory, tannins oxidise in insects at high gut pH to form semiquinone radicals and quinones as well as other reactive oxygen species capable of causing toxicity or oxidative stress in the gut tissues [5]. In herbivorous animals, tannins are believed to decrease protein utilisation, palatability and feed intake or interfere with the various enzyme activities.
Acorns, the fruit of oak trees (genus Quercus), are particularly rich in tannins and are commonly known to be an energy source for birds such as pigeons and woodpeckers, and many rodents including mice and squirrels. In horses, cattle and sheep, the ingestion of these tannin-rich nuts can cause severe loss of appetite, tremors and eventual death due to internal bleeding resulting from kidney or stomach damage. Pigs, however, seem to flourish on an acorn-rich diet. In fact, the ancient right of pannage, whereby local people can release their domestic pigs into forests for a certain period of time each year, is intended to remove the acorn crop so protecting foraging cattle and horses. This year in the New Forest, in southern England, the wet spring followed by the hot summer produced a bumper crop of acorns, resulting in the deaths of unusually high numbers of animals: forty-seven ponies and sixteen cattle had died by late November, compared with four to six animals in a normal year. As a result, the end of this year's pannage season, as decided by the Forestry Commission following consultation with the Court of Verderers, was extended twice – initially from the first week of November to mid-December and then further to the end of December [6, 7].
The image is a Creative Commons licensed picture shows a detail of a miniature, "Harvesting acorn to feed swine", from the Queen Mary Psalter (ca. 1310–1320).
Reference(s)
- Huang, X.D., Liang, J.B., Tan, H.Y., Yahya, R., Long, R. and Ho, Y.W. (2011) Protein-binding affinity of Leucaena condensed tannins of differing molecular weights. J. Agric. Food Chem., 59(19), 10677–10682.
- Geny, L., Saucier, C., Bracco, S., Daviaud, F. and Glories, Y. (2003) Composition and cellular localization of tannins in grape seeds during maturation. J. Agric. Food Chem., 51(27), 8051–8054.
- Lorrain, B., Ky, I., Pechamat, L. and Teissedre, P.L. (2013) Evolution of analysis of polyphenols from grapes, wines, and extracts. Molecules, 18(1), 1076–1100.
- Barbehenn, R.V. and Peter Constabel, C. (2011) Tannins in plant-herbivore interactions. Phytochemistry, 72(13), 1551–1565.
- Barbehenn, R.V., Jaros, A., Lee, G., Mozola, C., Weir, Q. and Salminen, J.P. (2009) Hydrolyzable tannins as "quantitative defenses": limited impact against Lymantria dispar caterpillars on hybrid poplar. J. Insect Physiol., 55(4), 297–304.
- Acorn glut kills New Forest ponies. news.bbc.co.uk. 11 November 2013.
- Pannage dates - further extension. Verderers of the New Forest.
2013

2nd December 2013, Dinitrogen oxide
The history of dinitrogen oxide (CHEBI:17045) involves some of the great names of 18thand 19th Century British science. First synthesised in 1772 by Joseph Priestley, who heated iron filings with a little nitric acid and isolated the resulting gas ('phlogisticated nitrous air'), it is better known today as 'laughing gas', nitrous oxide, or simply by its formula, N2O.In the 1790s, the English physician Thomas Beddoes, a proponent of 'pneumatic medicine', was administering various 'airs' to his patients, including oxygen, hydrogen, fixed air (carbon dioxide), and hydrocarbonate (a mixture of carbon monoxide and hydrogen). On the advice of Erasmus Darwin, the Scottish engineer James Watt called Beddoes in to treat his daughter, Jessie, who was suffering from consumption (now known as pulmonary tuberculosis) [1]. Although the treatment was unsuccessful (Jessie died, aged 15, in June 1794), Watt worked with Beddoes to publish the book 'Considerations on the Medicinal Use and on the Production of Factitious Airs' (1794), which included details of a new machine invented by Watt to produce the 'Factitious Airs' (i.e. nitrous oxide) as well as a novel 'breathing apparatus' for inhaling the gas. Watt also manufactured equipment to generate, clean and store gases for use in Beddoes' Pneumatic Institution at Hotwells in Bristol, to which Humphry Davy was appointed laboratory superintendent in 1798, aged just 19 [1].
It was due to Davy that nitrous oxide became the gas that was principally used at the Institution. He described the results of his experiments in his book 'Researches, chemical and philosophical, chiefly concerning nitrous oxide, or dephlogisticated nitrous air, and its respiration' (1800). Although he included notes on the analgesic effects of the gas (page 465) and suggested its use during surgical operations (page 556), over 40 years elapsed before attempts were made to use nitrous oxide for anaesthesia, for it was the bizarre, uncontrolled behaviour of the subjects who breathed the 'laughing gas' that attracted attention. 'Laughing gas parties' flourished among the British upper class, while Beddoes' hospital was rendered ridiculous. It was converted into a conventional hospital during a typhus outbreak in 1800 and closed in 1802; Davy left in 1801 to join Sir Joseph Banks at the newly-formed Royal Institution [1,2].
Although records of itinerant lecturers giving exhibitions of the effects of 'laughing gas' in the US date back to the 1830s, when Samuel Colt presented a show aimed at funding the patent for his revolver, it was not until 1844 that the anaesthetic properties of the gas were used for medical purposes. That it happened at all is due in no small part to the remarkable Gardner Quincy Colton (1814-1898). Born in Georgia, Vermont, the son of a weaver, he was apprenticed to a chairmaker for five years and subsequently moved to New York, making chairs there for several years. He enrolled in the Crosby Street College of Physicians and Surgeons (now Columbia University) in 1842, but left after 2 years, subsequently giving lectures on chemistry and natural philosophy. In the spring of 1844, he put on an exhibition of the properties of nitrous oxide in the Broadway Tabernacle, charging 25 cents each for tickets. Thousands of people attended – his total receipts came to $535, encouraging him to repeat his show throughout New England [3].
At a show in Hartford, Connecticut on December 10th 1844, Colton administered the gas to Samuel A. Cooley, a druggist's assistant. As Cooley subsequently danced and jumped about, he injured himself on some wooden benches on the stage. As the effect of the gas subsided, he took his seat next to a local dentist, Dr. Horace Wells, who observed that, despite having bleeding shins, Cooley did not notice any pain until the effects of the 'laughing gas' had worn off. At the end of the show, Wells discussed the matter with Colton and the following day, at Wells' office, Colton administered nitrous oxide to Wells while Dr. John B. Riggs, a neighbouring dentist, extracted one of Wells' teeth [4]. It was the first tooth ever drawn without pain. Wells subsequently used nitrous oxide anaesthesia for both dentistry and surgery, but after his death in 1848, use of nitrous oxide ceased. Its reintroduction in 1863 was due to Colton – he resumed his laughing gas exhibitions [5], through which he met and joined forces with another dentist, Joseph H. Smith of New Haven, Connecticut. By the time of Colton's death in 1898, it is estimated that he had extracted nearly a million teeth [6].
Over 200 years after Davy's experiments, nitrous oxide remains the most widely used inhalation anaesthetic in dentistry [7]. But while N2O exists naturally in the atmosphere in trace amounts (it is produced by bacteria in soils and oceans), the additional emissions resulting from human activity are now starting to cause problems. A new report by the United Nations Environment Programme (UNEP) warns that, following the adoption of the Montreal Protocol to phase out the production of CFCs and other ozone depleting substances, nitrous oxide is now the most important contributor to the loss of the ozone layer [8]. Furthermore, its ability to absorb energy combined with an average lifetime in the atmosphere of around 110 years means that it is a greenhouse gas with huge global warming potential – for a 100 year time horizon on a weight for weight basis, it is over 300 times more potent than carbon dioxide. Anthropogenic sources of nitrous oxide emissions are now the third largest climate-forcing agent, coming behind only carbon dioxide and methane. The report points out that concerted action to reduce emissions could therefore have a major impact on both the ozone layer and the rate of climate change. By far the largest single source of these emissions is agriculture, where the report recommends the more efficient use of fertilisers, minimising the loss of nitrogen to the environment during crop cultivation, and reducing food waste.
The background image is a Creative Commons licensed picture of a satirical cartoon of 1830 showing a Royal Institution lecture on pneumatics with Humphry Davy administering laughing gas while Count Rumford (who established the Royal Institution with Sir Joseph Banks) looks on.
Reference(s)
- Stansfield, D.A. and Stansfield, R.G. (1986) Dr. Thomas Beddoes and James Watt: preparatory work 1794-1796 for the Bristol Pneumatic Institute. Med. Hist., 30(3), 276–302.
- Wright, A.J. (1995) Davy comes to America: Woodhouse, Barton, and the nitrous oxide crossing. J. Clin. Anesth., 7(4), 348–355.
- Smith, G.B. (1991) Gardner Quincy Colton: pioneer of nitrous oxide anesthesia. Anesth. Analg., 72(6), 382–391.
- Haridas, R.P. (2013) Horace Wells' demonstration of nitrous oxide in Boston. Anesthesiology, 119(5), 1014–1022.
- Bause, G.S. (2009) Colton's laughing gas broadside. Anesthesiology, 110(3), 555.
- Gardner Q. Colton dead. New York Times, August 12th, 1898.
- Becker, D.E. and Rosenberg, M. (2008) Nitrous oxide and the inhalation anesthetics. Anesth. Prog., 55(4), 124–131.
- Drawing down N2O to protect climate and the ozone layer. United Nations Environment Programme, November 2013.
4th November 2013, N-[5-(1-naphthylmethyl)-1,3-thiazol-2-yl]cyclohexanecarboxamide
Systemic fungal infections or systemic mycoses have become a major concern over the past two decades owing to an increase in the proportion of immunodeficient people such as patients suffering from AIDS or neoplastic disease, recipients of hematopoietic stem cell and organ transplants, and premature infants often resulting in high morbidity and mortality rates [1]. In contrast to these opportunistic pathogens that cause diseases only in unhealthy hosts, the dimorphic fungi, which include Candida albicans [2], Coccidioides immitis [3] and Histoplasma capsulatum [4], are capable of infecting even immunocompetent individuals.Host cell toxicity has, however, impeded the development of antifungal agents to combat systemic mycoses. This is broadly related to the common eukaryotic nature of both the fungal pathogen and the mammalian host cell and the consequent difficulty in finding factors in the pathogen that are sufficiently distinctive from their host to be exploited as drug targets. The more commonly used clinical classes for the management of invasive mycoses include polyenes, azoles and echinocandins. The polyene amphotericin B, a bacterial metabolite, binds with the sterol components of the fungal membrane causing impairment of the membrane functions and subsequent cell death [5]. The azole antifungals such as fluconazole [6] inhibit the enzyme lanosterol 14α-demethylase involved in the synthesis of sterols, while the echinocandins, popularly known as the "penicillin of antifungals", inhibit the production of fungal cell wall glucan. Although the echinocandins are generally more effective antifungals than the polyenes and the azoles, they are ineffective against the Cryptococcus species and H. capsulatum [7].
The lung disease histoplasmosis is caused by inhaling spores of H. capsulatum. It is endemic to the Ohio and Mississippi river valleys, so giving rise to one of its alternative names Ohio valley disease. It is also known as Darling's disease after the name of its discoverer, Samuel Taylor Darling. The fungal pathogen grows as a saprobic filamentous mould in an environment associated with large amounts of bird or bat droppings. Contact with such soil can lead to a variety of clinical disorders varying from pulmonary infections that mimic symptoms of mild pneumonia to severe cases of respiratory distress syndrome in hosts exposed to large inocula of H. capsulatum [8].
In a recent phenotypic screening of a library of 3600 commercially available chemicals by Chad A. Rappleye and co-workers at The Ohio State University, a set of compounds was identified that exhibited antifungal activity against Histoplasma but which showed only low toxicity towards the host cells [9]. These compounds were based on purine analogue scaffolds and were simultaneously tested for inhibition of yeast growth and mammalian macrophage toxicity. The study identified an aminothiazole, N-[5-(1-naphthylmethyl)-1,3-thiazol-2-yl]cyclohexanecarboxamide (CHEBI:76002), better known as 41F5, that is highly toxic to the pathogen both in vitro and within macrophages but with reduced toxicity compared with other antifungals. Currently, the target of 41F5 is unidentified; however, its origin from a purinome-focused library (i.e. a library of proteins associated with purines) [10] suggests inhibitory activity against a purine-binding or purine-utilising factor in yeast cells. Although 41F5 bears structural resemblance to the recently licensed broad-spectrum antifungal abafungin [11], the former is distinct in being fungistatic (i.e. it inhibits the growth of fungi) thus suggesting a different antifungal cellular target.
The background image is a Creative Commons licensed picture showing two tuberculate macroconida (asexual, non-motile spores) of a Jamaican isolate of Histoplasma capsulatum.
Reference(s)
- Low, C.Y. and Rotstein, C. (2011) Emerging fungal infections in immunocompromised patients. F1000 Med. Rep., 3(14).
- Cascio, A., Pantaleo, D., Corona, G., Barberi, G., Delfino, D., Romeo, O., Iaria, C. and Barberi, I . (2013) Neonatal liver abscesses associated with candidemia. Three cases and review of literature. J. Matern.-Fetal Neonat. Med., doi: 10.3109/14767058.2013.837878, published online 2 Oct 2013.
- Brown, J., Benedict, K., Park, B.J. and Thompson G.R. III. (2013) Coccidioidomycosis: epidemiology. Clin. Epidemiol., 5(1), 185–197.
- Holbrook, E.D., Edwards, J.A., Youseff, B.H. and Rappleye, C.A. (2011) Definition of the extracellular proteome of pathogenic-phase Histoplasma capsulatum. J. Proteome Res., 10(4), 1929–1943.
- Charbonneau, C., Fournier, I., Dufresne, S., Barwicz, J. and Tancréde, P. (2001) The interactions of amphotericin B with various sterols in relation to its possible use in anticancer therapy. Biophys. Chem., 91(2), 125–133.
- Ouellet, D., Bramson, C., Roman, D., Remmers, A.E., Randinitis, E., Milton, A. and Gardner, M. (2007) Effects of three cytochrome P450 inhibitors, ketoconazole, fluconazole, and paroxetine, on the pharmacokinetics of lasofoxifene. Br. J. Clin. Pharmacol., 63(1), 59–66.
- Maligie, M.A and Selitrennikoff, C.P. (2005) Cryptococcus neoformans resistance to echinocandins: (1,3)β-glucan synthase activity is sensitive to echinocandins. Antimicrob. Agents Chemother., 49(7), 2851–2856.
- Kataria, Y.P., Campbell, P.B. and Burlingham, B.T. (1981) Acute pulmonary histoplasmosis presenting as adult respiratory distress syndrome: effect of therapy on clinical and laboratory features. South. Med. J., 74(5), 534–537, 542.
- Edwards, J.A., Kemski, M.M. and Rappleye, C.A. (2013) Identification of an aminothiazole with antifungal activity against intracellular Histoplasma capsulatum. Antimicrob. Agents Chemother., 57(9), 4349–4359.
- Fadden, P., Huang, K.H., Veal, J.M., Steed, P.M., Barabasz, A.F., Foley, B., Hu, M., Partridge, J.M., Rice, J., Scott, A., et al. (2010) Application of chemoproteomics to drug discovery: identification of a clinical candidate targeting hsp90. Chem. Biol., 17(7), 686–694.
- Tomillero, A. and Moral, M.A. (2010) Gateways to clinical trials. Methods Find. Exp. Clin. Pharmacol., 32(1), 47–86.
7th October 2013, Tramadol
The synthetic opioid analgesic tramadol (CHEBI:9648) was first marketed in 1977 by Grünenthal GmbH and has been used worldwide for the treatment of moderate to severe pain without any significant side-effects [1]. The drug has a wide range of additional applications, including treatment of rheumatoid arthritis, restless legs syndrome, motor neurone disease and fibromyalgia. In a recent research programme aimed at discovering natural products for use as pain relievers, a team led by Michel De Waard, a neuroscientist at the Université Joseph Fourier in Grenoble, has discovered the root bark of the African plant Nauclea latifolia to be a natural source of tramadol [2]. Although there are other previous examples of synthetic drugs that have later been found in nature, this is the first instance where the discovery has involved clinically relevant concentrations.Commonly known as the 'African peach' or 'pin cushion tree', N. latifolia is a flowering, sub-Saharan evergreen that grows widely across Central and West Africa and has a long tradition of use by local populations to treat a wide variety of ailments including epilepsy, malaria, general pain and many infectious diseases. To uncover the source of the plant's reported pain killing effect, the researchers fractioned methanolic extracts of the plant by high performance liquid chromatography (HPLC). After testing the resulting fractions in a biological assay using live mice, the team were able to determine the structure of the active compound in the fractions with the highest analgesic potency and confirm it to be identical with tramadol [2]. Further spectroscopic and isotope ratio analyses confirmed that the compound extracted was indeed natural in origin, and not a by-product of cross-contamination. The team have developed a simplified and inexpensive process for extracting the drug and are hopeful this might be of benefit to local communities. Although synthetic tramadol is not a costly drug in most developing countries, it is still considered expensive for many African citizens.
Both synthetic and the newly discovered natural tramadol are racemic mixtures composed of equal amounts of (1R,2R)-(+)- and (1S,2S)-(–)-enantiomers. Both enantiomers are pharmacologically active with (1R,2R)-(+)-tramadol(depicted in the image) exhibiting 10-fold higher analgesic activity compared with (1S,2S)-(–)-tramadol. In addition, the racemate seems to be superior to either enantiomer alone [3]. Relatively few natural products occur as racemic mixtures (less than 1% of the metabolome of the biosphere) [2]. The research team plan to investigate the biosynthetic pathway for the formation of the racemic mixture as well as the potential to extract tramadol from the other ten species of Nauclea that are known to exist in Africa.
The background image is a Creative Commons licensed picture showing the fruit and flower of Nauclea latifolia.
Reference(s)
- Leppert W. (2009). Tramadol as an analgesic for mild to moderate cancer pain. Pharmacol. Rep., 61, 978–992.
- Boumendjel, A., Taïwe, G.S., Bum, E.N., Chabrol, T., Beney, C., Sinniger, V., Haudecoeur, R., Marcourt, L., Challal, S., Queiroz, E.F., Souard, F., Le Borgne, M., Lomberget, T., Depaulis, A., Lavaud, C., Robins, R., Wolfender, J.-L., Bonaz, B. and De Waard., M. (2013). Occurrence of the synthetic analgesic tramadol in an African medicinal plant. Angew. Chem. Int. Ed., doi:10.1002/anie.201305697, published online 6 September 2013.
- Grond, S., Meuser, T., Zech, D., Hennig, U. and Lehmann, K.A. (1995). Analgesic efficacy and safety of tramadol enantiomers in comparison with the racemate: a randomised, double-blind study with gynaecological patients using intravenous patient-controlled analgesia. Pain, 62, 313–320.
2nd September 2013, Ivermectin
The avermectin antiparasitics were discovered as a result of a collaborative agreement between Merck & Co Inc, in the US and the Kitasato Institute (now part of Kitasato University) in Japan. From a batch of 54 microbial isolates sent from the Institute to Merck in March 1974, one proved to have particularly potent anthelminthic activity – crude extracts containing unknown amounts of the unknown active principle were active when incorporated into the diet of mice at a concentration of just 3 ppm. The active components were the avermectins, while the microorganism producing them was a new species of Streptomyces which was named S. avermitilis [1]. It is extremely rare – in the following five years, Merck screened 250,000 microbial cultures for activity against either Heligmosomoides bakeri or the model roundworm C. elegans; S. avermitilis was not found in any of the samples [2].As part of a chemical derivatisation programme, Merck found that the selective hydrogenation of the 22-23 double bond of a mixture of avermectin B1a and B1b gave a product with improved efficacy and safety characteristics. Now known as the broad-spectrum antiparasitic drug ivermectin (CHEBI:6078), it was found to be active against various nematodes occupying various segments of the intestinal tract in various species, against some extraintestinal parasites including blood-dwelling filarial microfilariae (the minute first-stage larvae), and against some internal parasites when applied externally [2]. Thanks to the development of ivermectin, there is now a real possibility that the parasitic disease onchocerciasis, the second most common infectious cause of blindness in the world, can be defeated.
Onchocerciasis affects large areas of sub-Saharan Africa as well as more isolated areas of Central and South America, and is one of only seventeen conditions now classed by the World Health Organisation (WHO) as a neglected tropical disease. In 2001, the WHO estimated that 18 million people were infected with the parasite [3]. The vectors for the disease are certain species of black fly of the genus Simulium [4], which are unusual in that they develop and breed in flowing water. Consequently, the disease is particularly prevalent in areas close to rivers, so giving rise to its alternative name, river blindness. Over 85 million people live in affected areas; in highly endemic areas, around 50% of men over the age of 40 are blind. As a consequence, large numbers of children miss out on education as they have to stay at home to act as full-time carers for parents who have become blind, while economic productivity in infected areas is greatly reduced, with vast tracts of arable land (usually the most fertile, being close to rivers) being abandoned, leaving only 'ghost villages' behind.
River blindness is caused by the bacterium Wolbachia pipientis, an endosymbiont of the parasitic roundworm Onchocerca volvulus which is transmitted to its ultimate human hosts via the bite of a female black fly that is infected by the roundworm. A black fly initially ingests microfilariae of the roundworm when it takes a blood meal from an infected human host. The microfilariae enter the gut and flight muscles of the fly and over the following week develop through the first, second, and third larval stages, whereupon they move to the fly's proboscis and into its saliva. When the black fly takes another blood meal, the larvae pass into the blood of the next human host. They then migrate to the subcutaneous tissue and undergo two more moults, forming nodules under the skin as they mature into adult worms over the next 6-12 months, whereupon they mate, after which the females will produce around 1,000 microfilariae each day. These can be found throughout the body of the human host, but preferentially reside in the skin (where they may be ingested when another black fly takes a blood meal from the human host, so continuing the cycle of infection) and the eye. When microfilariae die, surface proteins from the Wolbachia endosymbionts are released, inducing intense inflammatory immune responses and causing severe itching, depigmentation, swelling, and a loss of elasticity in the skin, together with inflammation of the cornea of the eye (keratitis). The resulting scarring slowly causes the entire cornea to become opaque, eventually leading to blindness.
Ivermectin is the drug of choice for onchocerciasis eradication programmes - it requires no refrigeration, and only needs to be administered once or twice a year. It rapidly kills the microfilariae and although it does not kill the adult worms, it does prevent the adult females from releasing microfilariae. Since the adult worms can live in their human hosts for many years, the treatment needs to be continued for the lifetime of the adult worms (up to ten years) to rid the human population of the worms and so break the cycle of infection.
Just such an eradication policy has been followed by Colombia, where the disease was restricted to the isolated riverside community of Naicioná. The Onchocerciasis Elimination Program of the Americas (OEPA), run by The Carter Center, organised the administration of ivermectin, donated by Merck, to the entire community every six months for 12 years. In 2007, it was found that the disease had stopped spreading. The drug treatment was stopped and the community was monitored for a further three years. In 2012, an international mission visited to verify that onchocerciasis was no longer present. On July 29th 2013, the WHO and the Colombian President Juan Manuel Santos officially announced that Colombia had become the first country ever to eliminate onchocerciasis. Ecuador has also completed the ivermectin treatment programme and has requested a visit from the WHO verification team, while Mexico and Guatemala (once the most affected areas of the American continent) have interrupted the cycle of the disease and suspended ivermectin treatment [5].
Although the problem of onchocerciasis in Africa is on a much greater scale to that on the American continent – 99% of those suffering from the disease live in Africa – it is thought that the methods used in Colombia will provide a useful model for combating the disease throughout the African continent.
The background image is a Creative Commons licensed picture showing an adult black fly (Simulium yahense) magnified approximately 100 times. In this remarkable image, obtained by conventional scanning electron microscopy, the parasitic roundworm Onchocerca volvulus can be seen emerging from the insect's antenna.
Reference(s)
- Kim, S.B. and Goodfellow, M. (2002) Streptomyces avermitilis sp. nov., nom. rev., a taxonomic home for the avermectin-producing streptomycetes. Int. J. Syst. Evol. Microbiol., 52(6), 2011–2014.
- Campbell, W.C. (2012) History of avermectin and ivermectin, with notes on the history of other macrocyclic lactone antiparasitic agents. Curr. Pharmaceut. Biotechnol., 13(6), 853–865.
- World Health Organisation (2001) http://www.who.int/water_sanitation_health/diseases/oncho/en/.
- Saint André, A.v., Blackwell, N.M., Hall, L.R., Hoerauf, A., Brattig, N.W., Volkmann, L., Taylor, M.J., Ford, L., Hise, A.G., Lass, J.H., Diaconu, E. and Pearlman, E. (2002) The role of endosymbiotic Wolbachia bacteria in the pathogenesis of river blindness. Science (New York, N.Y.), 295(5561), 1892–1895.
- Wyss, J. (2013) Colombia eradicates "river blindness," paving way for hemisphere and Africa. Miami Herald, http://www.who.int/water_sanitation_health/diseases/oncho/en/.
5th August 2013, Curcumin
Curcuma longa is a rhizomatous, herbaceous perennial member of the Zingiberaceae (ginger) family which is native to tropical South Asia. Its dried rhizome is the source of the spice turmeric, a dark yellow powder that has long been used as a flavouring and colouring agent in Indian and Middle Eastern cuisine as well as a therapeutic agent in Ayurvedic medicine for the treatment of both common ailments such as coughs, cold, cuts and burns and more serious diseases including cancer and Alzheimer's disease [1]. Among the several pharmacologically active metabolites [2] produced by the plant is curcumin (CHEBI:3962) [3], a hydrophobic polyphenol isolated from the rhizome, which has been shown to exhibit antioxidant [4], anti-inflammatory [5], antimicrobial [6], hepatoprotective [7] and anticancer [8,9] activities.Myeloma, also known as multiple myeloma (MM), is a type of bone marrow cancer in which abnormal white blood cells accumulate in the bone marrow and so interfere in the production of normal blood cells. Recently, in an effort to develop an effective cure for human MM, a team of Chinese researchers have adopted a 'hybrid molecule' strategy which aims to achieve a synergistic effect by combining drugs with similar pharmacological roles [10]. They synthesised a series of hybrid molecules based on the structures of curcumin [11] and thalidomide, a sedative, that was used as a sleeping pill and for the treatment of morning sickness during pregnancy but later withdrawn as it inadvertently became the most notable teratogen in history [12]. However, thalidomide has recently been identified as a potential anticancer agent [13].
Subsequent biological studies conducted against a series of MM cells established that the hybrids 5-[2-(feruloyl)ethen-1-yl]thalidomide (CHEBI:74774) and 5-(2,2-diferuloylethen-1-yl)thalidomide (CHEBI:74775) exhibited considerably enhanced lethal effects against the target human MM cells as compared to the individual molecules alone. The increased activity was attributed to the production of reactive oxygen species that trigger induction of apoptosis in cancer cells causing cell death. Furthermore, the researchers found that the hybrids also displayed NF-κB inhibitory activity against human lung cancer cells. These findings suggest that the hybrid strategy in drug design can yield novel multifunctional leads with improved activity as potential candidates for treatment of human MM.
Our background image is a Creative Commons licensed picture showing different parts of the plant Curcuma longa, published in Köhler's Medicinal Plants.
Reference(s)
- Singh, S. (2007) From exotic spice to modern drug? Cell, 130(5), 765–768.
- Dr. Duke's Phytochemical and Ethnobotanical Databases.
- Lechtenberg, M., Quandt, B. and Nahrstedt, A. (2004) Quantitative determination of curcuminoids in Curcuma rhizomes and rapid differentiation of Curcuma domestica Val. and Curcuma xanthorrhiza Roxb. by capillary electrophoresis. Phytochem. Anal., 15(3), 152–158.
- Kou, M.C., Chiou, S.Y., Weng, C.Y., Wang, L., Ho, C.T. and Wu, M.J. (2013) Curcuminoids distinctly exhibit antioxidant activities and regulate expression of scavenger receptors and heme oxygenase-1. Mol. Nutr. Food Res., doi: 10.1002/mnfr.201200227, published online 5 Feb 2013.
- Basnet, P. and Skalko-Basnet, N. (2011) Curcumin: an anti-inflammatory molecule from a curry spice on the path to cancer treatment. Molecules, 16(6), 4567–4598.
- De, R., Kundu, P., Swarnakar, S., Ramamurthy, T., Chowdhury, A., Nair, G.B. and Mukhopadhyay, A.K. (2009) Antimicrobial activity of curcumin against Helicobacter pylori isolates from India and during infections in mice. Antimicrob. Agents Chemother., 53(4), 1592–1597.
- Singh, M., Sasi, P., Gupta, V.H., Rai, G., Amarapurkar, D.N. and Wangikar, P.P. (2012) Protective effect of curcumin, silymarin and N-acetylcysteine on antitubercular drug-induced hepatotoxicity assessed in an in vitro model. Hum. Exp. Toxicol., 31(8), 788–797.
- Darvesh, A.S., Aggarwal, B.B. and Bishayee, A. (2012) Curcumin and liver cancer: a review. Curr. Pharm. Biotechnol., 13(1), 218–228.
- Ji, J.L, Huang, X.F. and Zhu, H.L. (2012) Curcumin and its formulations: potential anti-cancer agents. Anti-cancer Agents Med. Chem., 12(3), 210–218.
- Gediya, L.K. and Njar, V.C. (2009) Promise and challenges in drug discovery and development of hybrid anticancer drugs. Expert Opin. Drug Discovery, 4(11), 1099–1111.
- Liu, K., Zhang, D., Chojnacki, J., Du, Y., Fu, H., Grant, S. and Zhang, S. (2013) Design and biological characterization of hybrid compounds of curcumin and thalidomide for multiple myeloma. Org. Biomol. Chem., 11(29), 4757–4763.
- Ito, T., Ando, H. and Handa, H. (2011) Teratogenic effects of thalidomide: molecular mechanisms. Cell. Mol. Life Sci., 68(9), 1569–1579.
- Yang, G., Chen, W. and Wu, Y. (2013) Bortezomib, dexamethasone plus thalidomide for treatment of newly diagnosed multiple myeloma patients with or without renal impairment. Chin. J. Cancer Res. , 25(2), 155–160.

1st July 2013, Sulfamethazine
Since their discovery in the 1930s, sulfonamide antibiotics have been used to combat a wide spectrum of bacterial infections, including chlamydia, pneumonia, urinary tract infections and chronic ulcerative colitis. However, their side effects can include serious neurological problems such as nausea, headache, dizziness, hallucination and even psychosis. In addition, sulfonamide antibiotics can also cause allergic reactions ranging from various benign rashes to, in extreme cases, life-threatening Stevens-Johnson syndrome and toxic epidermal necrolysis. These side effects, along with growing bacterial resistance, have resulted in sulfonamide drugs being used much less frequently.Despite several decades of in-depth analysis of the sulfonamide family, the actual molecular mechanics behind the side effects have not been fully understood until a recent study headed by chemical biologist Kai Johnsson of the Swiss Federal Institute of Technology in Lausanne reported a possible cause of some of the neurological effects [1]. The researchers found that the arylsulfonamide moiety, present in all sulfonamide drugs, binds with the active site of an enzyme called sepiapterin reductase (SPR; EC 1.1.1.153), resulting in its inhibition [2]. This enzyme takes part in the biosynthesis of tetrahydrobiopterin, a compound essential for production of several neurotransmitters, including serotonin and dopamine. By using cell-based assays, the researchers showed that interference with the biosynthetic pathway eventually depletes the neurotransmitters, which in turn is thought to trigger the neurological side effects [3]. In addition, it was shown that one sulfonamide drug, sulfamethazine (CHEBI:102265), which is known not to have any side effects, was also a very weak inhibitor of SPR. This was attributed to the two methyl groups present on the pyrimidine ring making it more difficult for the molecule to fit in to the active site. In contrast, the analogue lacking the two methyl groups, sulfadiazine, was shown to be 370 times more potent as an SPR inhibitor. These findings open up the possibility of redesigning sulfonamide drugs so that they don't fit into the active site of SPR, or combining existing sulfonamide drugs with supplements to replace neurotransmitter molecules lost by inhibition of SPR.
The background image is a Creative Commons licensed picture showing the interior of an old pharmacy at the Kraków Museum of Pharmacy.
Reference(s)
- Haruki, H., Pedersen, M.G., Gorska, K.I., Pojer, F. and Johnsson, K. (2013) Tetrahydrobiopterin biosynthesis as an off-target of sulfa drugs. Science, 340, 987–991.
- Chidley, C., Haruki, H., Pedersen, M.G., Muller, E. and Johnsson, K. (2011) A yeast-based screen reveals that sulfasalazine inhibits tetrahydrobiopterin biosynthesis. Nat. Chem. Biol., 7, 375–383.
- Friedman, J., Roze, E., Abdenur, J.E., Chang, R., Gasperini, S., Saletti, V., Wali, G.M., Eiroa, H., Neville, B., Felice, A., et al. (2012) Sepiapterin reductase deficiency: a treatable mimic of cerebral palsy. Ann. Neurol., 71, 520–530.

3rd June 2013, GW 501516
This month's Entity of the Month is a failed drug candidate that has the rather unwieldy IUPAC name of {2-methyl-4-[({4-methyl-2-[4-(trifluoromethyl)phenyl]-1,3-thiazol-5-yl}methyl)sulfanyl]phenoxy} acetic acid. As the compound was never marketed, it has no brand or trade name and, as trials were stopped at an early stage, it also has no INN or US adopted name. Consequently, it is most commonly referred to in the primary literature by its research code [1], GW 501516 (CHEBI:73726), also GW 1516 and GSK-516.GW 501516 resulted from a research collaboration between SmithKline Beecham (now GlaxoSmithKline, GSK) and Ligand Pharmaceuticals that dates back to 1992. The compound was found to be a potent agonist of peroxisome proliferator-activated receptors type β/δ (PPARβ/δ), a type of nuclear receptor protein involved in the regulation of the metabolism of carbohydrates and lipids [2,3]. GSK began Phase I trials in 2001, hoping that GW 50156 could be used to stimulate muscles to burn fats and so be used as a treatment for hyperlipidaemia (high levels of lipids in the blood, often resulting from diabetes). In a trial in rats, the compound was found to increase fatty acid metabolism in skeletal muscle and give protection against diet-induced obesity and type 2 diabetes, while in obese rhesus monkeys increased levels of HDL ('good') cholesterol and lower levels of LDL ('bad') cholesterol were observed [4]. Ligand Pharmaceuticals received a $1 million 'milestone' payment from GSK in 2003 as Phase I development continued, while Phase II trials started in 2004. However, concurrent animal safety testing showed problems [5,6] and GSK stopped all development work on GW 501516 in 2006.
GW 501516 did not disappear without trace, however. The observation that giving high doses increased the exercise endurance of mice, together with the knowledge that (a) it could be easily synthesised [7] and (b) it was not controlled by sports doping regulations, made GW 501516 a potential performance-enhancing drug for athletes. Accordingly, the World Anti-Doping Agency (WADA) added PPARβ/δ modulators to their prohibited list in January 2009, and methods for detecting the use of GW 501516 in routine doping control tests (based on identification of the corresponding sulfoxide and sulfone, its urinary metabolites) were developed [8,9]. Nevertheless, WADA recently became aware that GW 501516 was being sold on the black market, commonly under the name 'endurobol', and in March of this year took the rare step of issuing an alert, pointing out to potential users that development of GW501516 was stopped when "serious toxicities" were discovered in pre-clinical studies [10]. The first positive test for 'endurobol' came within a month, with the announcement that the European track cycling champion Valery Kaykov had been sacked by his team, RusVelo [11]; several other professional cyclists have since tested positive [12,13].
So what are the "serious toxicities" referred to in the WADA alert? The initial human testing of GW 501516 was done using very low doses for short periods of time and did not show up any serious adverse effects. In contrast, animal testing used considerably higher doses over a much longer period. Rats given from 5 up to 40 mg/kg/day over a 2-year period developed cancers "in multiple tissues at all doses". Tissues affected included the stomach, liver, bladder, skin, thyroid, tongue, testes, ovaries and womb [5]. Similarly grim results were obtained from tests with mice [6]. For those athletes doping with GW 501516, there are clearly other things to worry about than simply being caught cheating.
The background image is a Creative Commons licensed picture showing protesters with a 'Tour de Doping' banner demonstrating against the widespread use of doping by competitors in the 2006 Tour de France.
Reference(s)
- Overington, J.P. (2013) The ontogeny and evolution of compound names. The ChEMBL-og - Open Data For Drug Discovery, February 3.
- Berger, J. and Moller, D.E. (2002) The mechanisms of action of PPARs. Annu. Rev. Med., 53, 409–435.
- Wang, Y.-X., Lee, C.-H., Tiep, S., Yu, R.T., Ham, J., Kang, H. and Evans, R.M. (2003) Peroxisome-proliferator-activated receptor δ activates fat metabolism to prevent obesity. Cell, 113(2), 159–170.
- Sprecher, D.L. (2007) Lipids, lipoproteins, and peroxisome proliferator activated receptor–δ. Am. J. Cardiol., 100(11, Suppl. 1), S20–S24.
- Geiger, L.E., Dunsford W.S., Lewis, D.J., Brennan, C., Liu, K.C. and Newsholme, S.J. (2009) Rat carcinogenicity study with GW50116, a PPAR delta agonist. The Toxicologist, Abstr. 48th Annu. Meet. Soc. Toxicol., Baltimore, March 15-19 2009, Abstr. 895.
- Newsholme, S.J., Dunsford W.S., Brodie, T., Brennan, C., Brown, M. and Geiger, L.E. (2009) Mouse carcinogenicity study with GW50116, a PPAR delta agonist. The Toxicologist, Abstr. 48th Annu. Meet. Soc. Toxicol., Baltimore, March 15-19 2009, Abstr. 896.
- Wei, Z.-L. and Kozikowski, A.P. (2003) A short and efficient synthesis of the pharmacological research tool GW501516 for the peroxisome proliferator-activated receptor δ. J. Org. Chem., 68(23), 9116–9118.
- Thevis, M., Moller, I., Thomas, A., Bueck, S., Rodchenkov, G., Bornatsch, W., Geyer, H. and Schanzer, W. (2010) Characterization ot two major urinary metabolites or the PPARδ-agonist GW1516 and implementation of the drug in routine doping controls. Anal. Bioanal. Chem., 396(7), 2479–2491.
- Thevis, M., Moller, I., Bueck, S. and Schanzer, W. (2013) Synthesis, mass spectrometric characterization, and analysis of the PPARδ agonist GW1516 and its major human metabolites: targets in sports drug testing. Methods Mol. Biol., 952, 301–312.
- WADA issues alert on GW501516. Play True magazine March 21 2013.
- Stokes, S. (2013) First cycling positive for GW501516, Rusvelo's Valery Kaykov provisionally suspended. Velonation News, April 11.
- Stokes, S. (2013) GW501516 positives confirmed, three of four riders are from same BCR Pizza Hut team. Velonation News, April 15.
- Stokes, S. (2013) Marlon Perez latest rider to test positive for GW501516. Velonation News, May 03.
7th May 2013, omacetaxine mepesuccinate
The plant alkaloid homoharringtonine (INN: omacetaxine mepesuccinate, CHEBI:71019), isolated from Cephalotaxus harringtonia (a slow-growing shrub or small tree, commonly known as the Japanese cow's tail pine, due to the shape of the leaves, or as the Japanese plum yew because of its plum-like fleshy fruits), was originally identified more than 35 years ago as a possible treatment for chronic myeloid leukaemia (CML, a cancer of the white blood cells accounting for up to a fifth of all cases of leukaemia in adults) and initial studies showed promising activity. However, the introduction of imatinib and related tyrosine kinase inhibitors (TKIs) halted the clinical development of homoharringtonine as a treatment for CML [1]. The advent of resistance to TKIs in some patients has led to renewed interest in homoharringtonine and in October 2012, the compound was approved by the US Food and Drug Administration (FDA) for treatment of CML in adults with resistance to established TKIs [2].Further potential for homoharringtonine in cancer treatment, this time in combination therapy with with tumour necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL), has been demonstrated in a recent publication by research groups headed by Petr Bartunek and Ladislav Andera at the Academy of Sciences of the Czech Republic in Prague [3]. TRAIL is under consideration as a potential anti-tumour agent. However, most primary human tumours are resistant to treatment with TRAIL alone, hence the need for combination therapy with drugs targeting the resistance. In high-throughput screening for novel agents that could sensitise TRAIL-resistant colorectal cancer cells to TRAIL-induced apoptosis (programmed cell death), the researchers found homoharringtonine to be a highly effective enhancer of TRAIL-mediated apoptosis of these resistant cells. The combination of TRAIL and homoharringtonine also led to the strong suppression of growth in tumours implanted into immunodeficient mice.
The background image is a Creative Commons licensed picture of Cephalotaxus harringtonia green fruits.
Reference(s)
- Wetzler, M. and Segal, D. (2011). Omacetaxine as an anticancer therapeutic: what is old is new again. Curr. Pharm. Des., 17, 59–64.
- FDA approves Synribo for chronic myelogenous leukemia (FDA press release), Oct 26, 2012.
- Beranova, L., Pombinho, A.R., Spegarova, J., Koc, M., Klanova, M., Molinsky, J., Klener, P., Bartunek, P. and Andera, L. (2013). The plant alkaloid and anti-leukemia drug homoharringtonine sensitizes resistant human colorectal carcinoma cells to TRAIL-induced apoptosis via multiple mechanisms. Apoptosis, 18, 739–750.
2nd April 2013, (–)-epigallocatechin 3-gallate
Alzheimer's disease (AD), as first described in 1906 by the German psychiatrist and neuropathologist Alois Alzheimer, is a progressive and fatal neurodegeneration of the brain manifested by symptoms of dementia and decline in cognition that escalate in severity as the disease advances, leading to impairment of everyday activities and neuropsychiatric symptoms [1–3].According to the amyloid cascade hypothesis for AD [4], the deposition or aggregation of the β-amyloid (Aβ) in the brain parenchyma is a central event in the disease pathology. The Aβ is a product of the cleavage of the amyloid precursor protein and has the natural propensity to aggregate and fold with a β-pleated sheet configuration and then stack on each other to form long fibrils and clumps known as senile plaques, in a process called cerebral amyloid angiopathy or congophilic angiopathy [5]. Additionally, elevated levels of transition metals such as Cu, Zn and Fe, essentially involved in homeostasis and proper neurological functioning of the brain, are also found in considerable concentrations in the amyloid plaque [6]. It is proposed that these metals have a relation to AD neuropathogenesis, namely in the aggregation of Aβ and the formation of reactive oxygen species which lead to oxidative stress and further neuronal death. Consequently, a number of potential therapeutic strategies to treat AD are based on the premise that utilizing novel bifunctional compounds that contain elements for metal chelation and Aβ interactions would provide anti-amyloidogenic effects [7,8].
According to a paper published recently by Mi Hee Lim and an interdisciplinary team of researchers at the University of Michigan, the extracts from green tea, Camellia sinensis, may block the formation of the Aβ plaques [9]. Their attention was drawn to (–)-epigallocatechin 3-gallate (EGCG, CHEBI:4806), a major catechin found in the leaves of the green tea, which has antioxidant [10], anti-cancer [11], anti-HIV [12] and neuroprotective activities [13]. The authors propose that not only does EGCG prevent the formation of aggregates but that it also breaks down existing aggregate structures in the proteins that contained metals – specifically copper, iron and zinc. The studies also suggest that the overall reactivity of EGCG with a metal-free or metal-associated Aβ species is associated with its ability to influence formation of the compact conformation of Aβ and ternary complexes containing Aβ, Cu or Zn and EGCG in multiple stoichiometries. The study essentially aims at building a structure-based mechanism for EGCG action towards Aβ and builds a platform for development and application of small molecules as therapeutic agents for AD.
The background image is a Creative Commons licensed picture showing a Chinese teapot.
Reference(s)
- Berchtold, N.C. and Cotman, C.W. (1998) Evolution in the conceptualization of dementia and Alzheimer�s disease: Greco-Roman period to the 1960s. Neurobiol. Aging, 19(3), 173–189.
- Boller, F. and Forbes, M.M. (1998) History of dementia and dementia in history: An overview. J. Neurol. Sci., 158(2), 125–133.
- Tran, M., Bédard, M., Dubois, S., Weaver, B. and Molloy, D.W. (2013) The influences of psychotic symptoms on the activities of daily living of individuals with Alzheimer disease: a longitudinal analysis. Aging Ment. Health, 17, in press.
- Karran, E., Mercken, M. and De Strooper, B. (2011) The amyloid cascade hypothesis for Alzheimer�s disease: an appraisal for the development of therapeutics. Nat. Rev. Drug. Discov., 10(9), 698–712.
- Weller, R.O., Preston, S.D., Subash, M. and Carare, R.O. (2009) Cerebral amyloid angiopathy in the aetiology and immunotherapy of Alzheimer disease. Alzheimers Res. Ther., 1(2), 6.
- Pithadia, A.S. and Lim, M.H (2012) Metal-associated amyloid-β species in Alzheimer�s disease. Curr. Opin. Chem. Biol., 16(1–2), 67–73.
- Wu, W.H., Lei, P., Liu, Q., Hu, J., Gunn, A.P., Chen, M.S., Rui, Y.F., Su, X.Y., Xie, Z.P., Zhao, Y.F., Bush, A.I. and Li, Y.M. (2008) Sequestration of copper from &beta-amyloid promotes selective lysis by cyclen-hybrid cleavage agents. J. Biol. Chem., 283(46), 31657–31664.
- Ono, K., Hasegawa, K., Naiki, H. and Yamada, M. (2004) Curcumin has potent anti-amyloidogenic effects for Alzheimer's β-amyloid fibrils in vitro. J. Neurosci. Res., 75(6), 742–750.
- Hyung, S.J., Detoma, A.S., Brender, J.R., Lee, S., Vivekanandan, S., Kochi, A., Choi, J.S., Ramamoorthy, A., Ruotolo, B.T. and Lim, M.H. (2013) Insights into antiamyloidogenic properties of the green tea extract (–)-epigallocatechin-3-gallate toward metal–associated amyloid-β species. Proc. Natl. Acad. Sci. U. S. A., 110(10), 3743–3748.
- Wang, Y., Zhao, Y., Andrae-Marobela, K., Okatch, H. and Xiao, J. (2013) Tea polysaccharides as food antioxidants: An old woman's tale? Food Chem., 138(2–3), 1923–1927.
- Fang, M.Z., Wang, Y., Ai, N., Hou, Z., Sun, Y., Lu, H., Welsh, W. and Yang, C.S. (2003) Tea polyphenol (–)-epigallocatechin-3-gallate inhibits DNA methyltransferase and reactivates methylation-silenced genes in cancer cell lines. Cancer Res., 63(22), 7563–7570.
- Williamson, M.P., McCormick, T.G., Nance, C.L and Shearer, W.T. (2006) Epigallocatechin gallate, the main polyphenol in green tea, binds to the T-cell receptor, CD4: Potential for HIV-1 therapy. J. Allergy Clin. Immunol., 118(6), 1369–1374.
- Itoh, T., Imano, M., Nishida, S., Tsubaki, M., Mizuguchi, N., Hashimoto, S., Ito, A. and Satou, T. (2012) (–)-Epigallocatechin-3-gallate increases the number of neural stem cells around the damaged area after rat traumatic brain injury. J. Neural Transm., 119(8), 877–890.
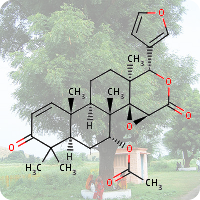
4th March 2013, gedunin
The Indian neem tree, Azadirachta indica, is a quick-growing evergreen member of the Meliaceae (mahogany) family which is native to the Indian subcontinent and has been introduced to many other areas in the tropics. Also known as known as 'Heal all' and 'Nature's pharmacy', it has long been used in traditional Indian medicine. Among the pharmacologically active metabolites produced by the tree is gedunin (CHEBI:65954) [1,2], a pentacyclic tetranortriterpenoid isolated from the bark, which has been shown to possess antimalarial [3], insecticidal [4], and anticancer activity [5]. The latter has been shown to result from the inhibition of the 90 kDa heat shock protein (Hsp90) [6,7].Hsp90 is among the most important of chaperone proteins (proteins that help other proteins to fold and unfold properly): it has been estimated that the Hsp90 protein-foldingmachine may handle up to 10% or all cellular proteins [8]. Furthermore, it is required for the stability and function of a number of conditionally activiated signalling proteins and mutated proteins that promote cancer cell growth and survival [9]. Consequently, inhibiting the action of Hsp90 has become an important strategy for the development of novel cancer treatments in recent years [10] and a number of Hsp90 inhibitors are in clinical trials for cancer treatment [11–14]. However, the clinical efficacy of most Hsp90 inhibitors has been disappointing for, in addition to inhibiting Hsp90, the compounds have inadvertently caused the overexpression of Hsp70 and Hsp27, which protect cancer cells from apoptosis (programmed cell death), and this has reduced the benefit of inhibiting the action of Hsp90.
Now a group of scientists led by Ahmed Chadli at Georgia Regents University Cancer Center have found that gedunin inhibits Hsp90 indirectly [15]. Hsp90 works in combination with a number of chaparones and co-chaperones (helper proteins). Chadli's group has shown that gedunin binds directly to a co-chaperone of Hsp90 called p23, resulting in the Hsp90 machine being deactivated without the undesired production of the anti-apoptotic proteins.
Their discovery opens up new possibilities for cancer treatments, in which Hsp90 is targeted indirectly through targeting its co-chaperones; over twenty co-chaperone proteins have so far been shown to regulate the Hsp90 machine. It is thought that their findings will be particularly relevant for future drug development for hormone-dependent cancers, including breast, prostate, and endometrial cancers.
The background image is a Creative Commons licensed picture showing a neem tree at Rawanjana Dungar railway station, Rajastan.
Reference(s)
- Akisanya, A., Bevan, C.W.L., Hirst, J., Halsall, T.G. and Taylor, D.A.H. (1960) Petroleum extracts from the genus Entandrophragma. J. Chem. Soc., 3827–3829.
- Akisanya, A., Bevan, C.W.L., Halsall, T.G., Powell, J.W. and Taylor, D.A.H. (1961) Some reactions of gedunin. J. Chem. Soc., 3705–3708.
- Khalid, S.A., Duddeck, H. and Gonzalez-Sierra, M. (1989) Isolation and characterization of an antimalarial agent of the neem tree Azadirachta indica. J. Nat. Prod., 52(5), 922–926.
- Nathan, S.S., Kalaivani, K., Chung, P.G. and Murugan, K. (2006) Effect of neem limonoids on lactate dehydrogenase (LDH) of the rice leaffolder, Cnaphalocrocis medinalis (Guenée) (Insecta: Lepidoptera: Pyralidae). Chemosphere, 62(8), 1388–1393 .
- Uddin, S.J., Nahar, L., Shilpi, J.A., Shoeb, M., Borkowski, T., Gibbons, S., Middleton, M., Byres, M. and Sarker, S.D. (2007) Gedunin, a limonoid from Xylocarpus granatum, inhibits the growth of CaCo-2 colon cancer cell line in vitro. Phytother. Res., 21(8), 757–761.
- Lamb, J., Crawford, E.D., Peck, D., Modell, J.W., Blat, I.C., Wrobel, M.J., Lerner, J., Brunet, J.P., Subramanian, A., Ross, K.N., Reich, M., Hieronymus, H., Wei, G. and Carr, S.A. (2006) The connectivity map: using gene-expression signatures to connect small molecules, genes, and disease. Science (Washington, DC, U. S.), 313, 1929–1935.
- Hieronymus, H., Lamb, J., Ross, K.N., Peng, X.P., Clement, C., Rodina, A., Nieto, M., Du, J., Stegmaier, K., Raj, S.M., Maloney, K.N., Clardy, J., Hahn, W.C., Chiosis, G. and Golub, T.R. (2006) Gene expression signature-based chemical genomic prediction identifies a novel class of HSP90 pathway modulators. Cancer Cell, 10(4), 321–330.
- Picard, D. (2012) Preface to Hsp90. Biochim. Biophys. Acta, Mol. Cell Res., 1823(3), 605–606.
- Neckers, L. (2007) Heat shock protein 90: the cancer chaperone. J. Biosci, 32(3), 517–530.
- Sidera, K. and Patsavoudi, E. (2013) HSP90 inhibitors: current development and potential in cancer therapy. Recent Pat. Anti-Cancer Drug Discovery, 8, in press.
- Blagg, B.S. and Kerr, T.D. (2006) Hsp90 inhibitors: small molecules that transform the Hsp90 protein folding machinery into a catalyst for protein degradation. Med. Res. Rev., 26(3), 310–338.
- Xiao, L., Lu, X. and Ruden, D.M. (2006) Effectiveness of Hsp90 inhibitors as anti-cancer drugs. Mini-Rev. Med. Chem., 6(10), 1137–1143.
- Kim, Y.S., Alarcon, S.V., Lee, S., Lee, M.J., Giaccone, G., Neckers, L. and Trepel, J.B. (2009) Update on Hsp90 inhibitors in clinical trial. Curr. Top. Med. Chem., 9(15), 1479–1492.
- Trepel, J., Mollapour, M., Giaccone, G. and Neckers, L. (2010) Targeting the dynamic HSP90 complex in cancer. Nat. Rev. Cancer, 10(8), 537–549.
- Patwardhan, C.A., Fauq, A., Peterson, L.B., Miller, C., Blagg, B.S. and Chadli, A. (2013) Gedunin inactivates the co-chaperone p23 causing cancer cell death by apoptosis. J. Biol. Chem., 288, in press.
4th February 2013, caffeine
In recognition of the copious amounts of caffeine consumed by various members of the ChEBI team throughout the first 99 releases, caffeine (CHEBI:27732) has been selected as our entity of the month to commemorate the 100th release of ChEBI.Caffeine is the common name for 1,3,7-trimethylxanthine. The word "caffeine" originates from the German "kaffee" and the French "café", each meaning coffee. In purified form, caffeine exists as a white powder with a distinctive bitter flavour. Caffeine is naturally found in certain leaves, beans, and fruits of over 60 plants worldwide, where it acts as a natural pesticide. Caffeine is synthesised in plants from the purine nucleotides AMP, GMP, and IMP. These in turn are transformed into xanthosine, then theobromine and eventually caffeine [1]. In humans, caffeine acts as a central nervous system stimulant, temporarily warding off drowsiness and enhancing alertness. The most common sources of caffeine in the human diet are coffee, tea, soft drinks, energy drinks and chocolate derived from cocoa beans [2].
The long term effects of moderate caffeine consumption (up to 300 mg per day, equivalent to about three cups of coffee) are thought to be a reduction in the risk of developing Parkinson's disease, type 2 diabetes, hepatic diseases, cardiovascular disease and some forms of cancer. However, heavy consumption (more than 500-600 mg per day) may have negative effects in some users, such as insomnia, anxiety, mood swings, nausea, palpitations, headaches and muscle tremors. Fatal caffeine overdoses in adults are relatively rare and require the ingestion of a large quantity of the drug, typically in excess of 5 g [3].
Two recent reports have served to highlight both the positive and negative effects of caffeine consumption. Ronald Postuma of McGill University in Montreal, Canada, and co-workers have recently studied the effects of regular caffeine consumption on patients who already have the symptoms of Parkinson's disease. They gave 61 people with the disease a six-week course of pills containing the caffeine equivalent of about three cups of coffee every day, or a placebo. Only people in the caffeine group showed significant improvement in tests for motor problems, such as the severity of tremors, and general mobility [4].
Unlike many other psychoactive substances, caffeine is both legal and unregulated in nearly all parts of the world. Energy drinks, which at present are not regulated as they are classed as nutritional supplements, have been under scrutiny because of their high caffeine content. In October 2012, parents from Maryland filed a wrongful death lawsuit against an energy drink manufacturer after their 14-year-old daughter died of cardiac arrest. She had reportedly consumed two 24-ounce cans of the beverage within a 24-hour period. It is thought that she suffered from an hereditary heart condition which may have been triggered by the sudden rush of caffeine [5]. In November 2012, The US Food and Drug Administration (FDA) released incident reports describing several deaths that may have occurred following the consumption of energy drinks. The filing of such reports with the FDA does not prove that a product was directly responsible for a death or an injury. In a released statement, the FDA stated that it was likely to seek advice from outside experts to help determine whether energy drinks posed particular risks to teenagers or people with underlying health problems [5]. In Canada, an independent panel made several recommendations, including that such beverages are labelled as �stimulant drug-containing drinks� and new regulations have been put in place to limit caffeine levels in energy drinks to 180 mg [6].
Reference(s)
- Ashihara, H., Monteiro, A.M., Gillies, F.M. and Crozier, A. (1996). Biosynthesis of caffeine in leaves of coffee. Plant Physiol., 111, 747–753.
- http://www.howstuffworks.com/caffeine.htm
- Kerrigan, S. and Lindsey T. (2005). Fatal caffeine overdose: two case reports. Forensic Sci. Int., 153, 67–9.
- Postuma, R.B., Lang A.E., Munhoz, R.P., Charland, K., Pelletier, A., Moscovich, M., Filla L., Zanatta, D., Romenets, S.R., Altman, R., Chuang, R. and Shah, B. (2012). Caffeine for treatment of Parkinson disease. A randomized controlled trial. Neurology, 79, 651–659.
- http://features.blogs.fortune.cnn.com/2013/01/23/energy-drinks-brace-for-caffeine-crash/
- http://www.hc-sc.gc.ca/fn-an/legislation/pol/energy-drinks-boissons-energisantes-eng.php
7th January 2013, Acrylamide
Acrylamide (CHEBI:28619) is an industrially produced reactive molecule widely used as a chemical intermediate in the preparation of polymers such as polyacrylamide,a polymer which can be synthesised in both linear-chain and cross-linked forms and is utilized in many processes including the synthesis of dyes, the preparation of copolymers for contact lenses and in sealant grouts in the construction of dam foundations, tunnels and sewers. However, despite its widespread utility, acrylamide has been shown to be a potent neurotoxin in both humans and animals. Acrylamide also acts as a reproductive toxin, genotoxin and carcinogen in rodents [1]. In addition, based on several cohort studies of cancers in humans with potential for occupational exposure to acrylamide, the chemical is also classified as 'probably carcinogenic to humans' (class 2A) by the International Agency for Research on Cancer [2].Acrylamide toxicity became a major concern when a Swedish study announced the discovery of considerable levels of the compound in starch-based foods such as potato crisps, french fries, bread and other foodstuffs. Acrylamide is formed during high-temperature cooking via the Maillard reaction involving the reaction of a reducing sugar with the amino acid L-asparagine [3]. However a link between the dietary intake of acrylamide and cancer could not be confirmed until a Dutch epidemiological survey, involving around 62,000 women over a period of 11 years, concluded that women who ingested 40 �g per day of acrylamide via their diet were twice as likely to develop womb or ovarian cancer than women who had eaten only 9 �g per day [4]. This has evoked several research efforts aimed at reducing the dietary levels of the chemical in foodstuffs through new farming and food-processing techniques.
One of the ways to reduce acrylamide formation in potatoes is by developing new cultivars that are low in the acrylamide precursors – L-asparagine and the reducing sugars. As a part of these efforts, a team of researchers led by Nigel Halford from Rothamsted Research, UK, measured the levels of acrylamide precursors in nine different varieties of potatoes along with the amount of acrylamide formed during frying. The precursor–acrylamide relationship was found to be dependent on the strain of the potato used [5].
The study proposed that the two crisp-potato varieties Lady Clare and Saturna showed acrylamide formation that was consistently below the European Commission's ideal of 1000 ppb. However the complexity of the precursor–acrylamide relationships and the sensitivity of the potatoes to seasonal variations make it difficult to identify specific targets for breeding programmes. Nevertheless, researchers continue to aim at developing more efficient tools to produce low-acrylamide varieties of potatoes to comply with possible harsher regulatory limits.
The background image is that of potatoes frying in oil, during which process the Maillard reaction gives rise to acrylamide.
Reference(s)
- Parzefall, W. (2008) Minireview on the toxicity of dietary acrylamide. Food Chem. Toxicol., 46(4), 1360–1364.
- IARC Monograph (1994) Acrylamide. IARC Monogr. Eval. Carcinog. Risks Hum., 60, 389–433.
- Svensson, K., Abramsson, L., Becker, W., Glynn, A., Hellen�s, K.-E., Lind, Y. and Ros�n, J. (2003) Dietary intake of acrylamide in Sweden. Food Chem. Toxicol., 41(11), 1581–1586.
- Hogervorst, J. G., Schouten, L. J., Konings, E. J., Goldbohm, R. A. and van den Brandt, P. A. (2007) A prospective study of dietary acrylamide intake and the risk of endometrial, ovarian, and breast cancer.Cancer Epidemiol., Biomarkers Prev., 16, 2304–2313.
- Halford, N. G., Muttucumaru, N., Powers S. J., Gillatt, P. N., Hartley, L., Elmore, J. S. and Mottram, D. S. (2012) Concentrations of free amino acids and sugars in nine potato varieties: effects of storage and relationship with acrylamide formation. J. Agric. Food Chem., 60(48), 12044–12055.
2012

3rd December 2012, Antibiotic
This month's entity is not an individual compound, but rather a class of compounds. So what is an antibiotic? One of the most quoted of the early definitions was published by Selman A. Waksman (the co-discoverer of streptomycin) in 1947. His definition was 117 words in length but, in short, 'antibiotic' was described as any substance produced by a microorganism that killed or inhibited the growth of bacteria or other microorganisms [1]. Most published definitions still limit the meaning of 'antibiotic' to microbial natural products [2], despite the fact that most scientists find the distinction 'rather academic' [3]. The term 'antibiotic' is now commonly used to include semisynthetic modifications of natural compounds, such as many of the penicillins and cephalosporins, as well as compounds produced entirely by chemical synthesis, for example the sulfonamides.The medical profession has now had an extensive arsenal of antibiotics available for the treatment of infectious diseases for well over 50 years. Consequently, it has become difficult to envisage just how rapidly microorganisms could overwhelm the body's defences and just how precarious life could be before antibiotics became available. A perfectly healthy individual could die of septicaemia from a simple cut, a case in point being that of Calvin Coolidge Jr., the youngest son of the 30th President of the United States who, during the afternoon of June 30th 1924, while playing tennis with his older brother, developed a blister on his right foot. Septicaemia developed, and the best medical practitioners in the nation's capital couldn't save him; he died aged 16 at the Walter Reed Army General Hospital just seven days later [4]. Many other bacterial infections, most notably tuberculosis, were also routine killers.
The introduction of the first sulfonamides (in 1935) and, particularly, penicillins (in 1940) caused the mortality rate associated with bacterial infections to plummet. However, the power of these new antibiotics soon began to wane owing to the ability of bacteria to develop resistance to them.
Alexander Fleming, the discoverer of penicillin, warned against misusing antibiotics at the end of his acceptance speech for the 1945 Nobel Prize for Physiology or Medicine [5]. "The time may come when penicillin can be bought by anyone in the shops. Then there is the danger that the ignorant man may easily underdose himself and by exposing his microbes to non-lethal quantities of the drug make them resistant." The warning has been repeated regularly over the years; for example the Alliance for the Prudent Use of Antibiotics (APUA), a non-profit group based at Tufts University Medical School, has been promoting appropriate antibiotic use to contain drug resistance for over thirty years. Despite this, the warnings have been largely ignored.
For many years, the problem was masked by a seemingly endless supply of new and better antibiotics from the major pharmaceutical companies. Thus between 1940 and 1962, more than 20 new classes of antibiotics were marketed, so that when bacteria developed resistance to one antibiotic there was always another one available to take its place [6]. Since 1962, however, only four new classes have entered the market, while the number of large pharmaceutical companies engaged in the discovery of antibiotics has fallen markedly. It typically takes only two years between the introduction of a new antibiotic and the observance of resistance to it, but the process of developing a new antibiotic is long (around ten years) and expensive (typically over $300 million). Combined with the emergence of multi-drug-resistant bacteria, the result is that insufficient new classes of antibiotics or new members of existing classes are reaching the market to keep pace with the development of resistant bacteria. The need for antibiotics targeting Gram-negative bacteria is particularly acute.
In cases of severe infections, antibiotic treatment has to start 'blind' until the infecting bacteria have been identified in the laboratory, which can take two days. As bacterial resistance increases, so does the likelihood that the 'blind' treatment will not work. Where there is blood poisoning (sepsis) this delay in fighting the infection doubles the risk of death. Doctors are increasingly having to use carbapenems, the 'reserve' antibiotics kept for use when other treatments have failed. Now the spread of resistance to carbapenems is being seen as well.
While it is still almost always possible to find an antibiotic to treat any bacterial infection, the situation is far from ideal. Alternative antibiotics are often more expensive than the 'normal' treatments and can take much longer to work, as they are often not as good at killing the bacteria; they can also cause serious side effects. Thus the cost of treating one person suffering from multi-drug-resistant tuberculosis (MDR-TB) can be as much as 200 times that of treating someone with the non-resistant variety. The burden of this cost is likely to fall disproportionately on poor countries - of the 310,000 cases of MDR-TB reported in the world in 2011, almost 60% were in India, China or the Russian Federation [7].
As part of an on-going effort to reduce the unnecessary use of antibiotics and so cut down the development and spread of resistance, the U.K. Health Protection Agency (HPA) chose 18th November 2012, European Antibiotics Awareness Day (EAAD), to urge people to think twice before they ask for antibiotics from their medical practitioners to treat cold and flu symptoms - colds and influenza are viral infections, while antibiotics are active against bacteria, not viruses [8]. However, this is only one aspect of a much bigger problem.
In many parts of the world, antibiotics can be purchased 'over the counter', without the need for any diagnosis or recommendations for proper use. Antibiotics are also readily available via the internet, where effective regulation to limit supplies to cases where they are needed is virtually impossible, increasing still further the number of human incubators from which resistant strains of bacteria can evolve. For many years, antibiotics were routinely given to many farm animals, not because they were sick, but simply to promote growth. Denmark, the world's biggest exporter of pork, began phasing out the practice more than 15 years ago, and an EU-wide ban was introduced at the start of 2006; however, the practice continues elsewhere, including in the United States. Congresswoman Louise Slaughter, the only microbiologist in Congress, has calculated from FDA figures that up to 80% of all of the antibiotics used in the U.S. are given to livestock, mostly as growth promotion agents.
Our image is a Creative Commons licensed picture of a Faroe Islands postage stamp issued in 1983, depicting Sir Alexander Fleming.
Reference(s)
- Waksman, S.A. (1947) What is an antibiotic or antibiotic substance? Mycologia, 39, 565–569.
- Bentley, R. and Bennett, J.W. (2003) What is an antibiotic? Revisited. Adv. Appl. Microbiol., 52, 303–331.
- Pratt, W.B. and Fekety, R. (1986) "The antimicrobial drugs." Oxford University Press, New York.
- President's son, Calvin Jr., 16, dies as parents watch. New York Times, July 8th, 1924.
- http://www.nobelprize.org/nobel_prizes/medicine/laureates/1945/fleming-lecture.pdf
- Coates, A.R., Halls, G. and Hyu, Y. (2011) Novel classes of antibiotics or more of the same? Br. J. Pharmacol, 163(1), 184–194.
- Tuberculosis. World Health Organisation Factsheet 104 (October 2012).
- Antibiotics and you. Health Protection Agency Press Release, November 14th 2012.

5th November 2012, dinitrogen
Having part of one's stomach removed is perhaps not the way one would celebrate a coming of age. Nevertheless, owing to the misuse of liquid nitrogen (CHEBI:17997), this was the fate of British teenager Gaby Scanlon, who suffered a perforated stomach after drinking a cocktail prepared with liquid nitrogen [1]. Scanlon, who was out with friends in Lancaster on her 18th birthday, was reported to have become breathless and developed severe stomach pains before being taken to the Royal Lancaster Infirmary for life-saving surgery. The procedure, a gastrectomy, involves connecting the oesophagus to the small intestine. As a result Scanlon is likely to have to alter her diet, such as by eating smaller meals more frequently and taking multivitamin supplements.Liquid nitrogen is a cryogenic fluid which boils at -196°C at atmospheric pressure and starts to evaporate the moment it comes into contact with room temperature air, creating a dramatic dry-ice effect caused by condensation of water vapour in the air. But if the nitrogen has not boiled away fully, as a liquid it has the power to freeze objects in a matter of seconds. If swallowed, liquid nitrogen can cause cold burns to the mouth, throat and stomach, killing the tissue. As the frozen vapour hits the stomach it rapidly warms, releasing large volumes of air which can burst the stomach. As a result of the accident, the Food Standards Agency has issued a warning about the dangers of liquid nitrogen [2] and David Morris, the Member of Parliament for the constituency of Morecambe and Lunesdale, has called on the UK Parliament to ban liquid nitrogen cocktails [3].
Reference(s)
1st October 2012, everolimus
Over the last few decades, major improvements have been achieved in the management of breast cancer, mirrored by a significant decrease in the mortality rate despite an increasing prevalence of the disease. However, metastatic breast cancer (also known as secondary breast cancer) remains common and is the cause of death in nearly 12,000 women annually in the UK [1]. It occurs when the breast cancer cells spread from the first primary tumour in the breast to another distant part of the body (metastasis) which happens through the blood stream or lymphatic system. This type of spread is also referred to as advanced breast cancer or stage 4 cancer.Adjuvant endocrine therapy [2] that employs the use of a selective estrogen receptor antagonist such as tamoxifen or an aromatase inhibitor has been a cornerstone of treatment for patients diagnosed with metastatic breast cancer in recent years. However, patients generally tend to develop resistance to such hormone therapy. Apparently, the mechanism for this resistance is over-activation of the mTOR (mammalian transporter of rapamycin) pathway which regulates cell growth, proliferation, motility and survival and is over-active in many forms of cancer [3].
At the American Society of Clinical Oncology (ASCO) Breast Cancer Symposium held last month in San Francisco, Prof. Hope Rugo, Director of Breast Oncology and Clinical Trials Education and Professor of Medicine at the UCSF Helen Diller Family Comprehensive Cancer Center in San Francisco, presented the updated 18-month results of the pivotal phase III Breast Cancer Trials of Oral Everolimus (BOLERO-2) [4]. The trials indicate that treatment with everolimus (CHEBI:68478), a derivative of sirolimus, used in combination with the aromatase inhibitor exemestane, extended median progression-free survival in patients to 7.8 months compared with 3.2 months for the control group treated with exemestane alone. These progression-free survival benefits were stable for all the subgroups studied including age and geography.
Everolimus acts as an mTOR inhibitor, modulating the progression of the tumour. Given the positive results, everolimus has entered National Comprehensive Cancer Network (NCCN) 2012 guidelines and has been approved as a drug for advanced breast cancer by the FDA [5].
We have chosen everolimus as our entity of month in support of Macmillan Cancer Support, one of the largest cancer support charities in the UK, who have recently organised the World�s Biggest Coffee Morning, a fundraising event spread over numerous locations in the UK, including our Hinxton Campus.
The background image is a Creative Commons licensed picture of a giant pink ribbon in a market in Louisville, Kentucky, for Breast Cancer awareness.
Reference(s)
- Coleman, R.E., Bertelli, G., Beaumont, T., Kunkler, I., Miles, D., Simmonds, P.D., Jones, A.L. and Smith, I.E. (2011) UK guidance document: Treatment of metastatic breast cancer. Clin. Oncol., 24(3), 169–176.
- Smith, I.E. and Dowsett, M.(2003) Aromatase inhibitors in breast cancer. N. Engl. J. Med., 348(24), 2431–2442.
- (2012) Everolimus approved for HR-positive breast cancer. Cancer Discovery, 2(9), 756.
- Azvolinsky A. (2012) Everolimus for advanced breast cancer: An update from the BOLERO-2 Trial. CancerNetwork, September 18.
- FDA approves Afinitor for advanced breast cancer (FDA press release), July 20, 2012.

3rd September 2012, β-Carotene
Vitamin A deficiency is of public health significance in the developing world. Approximately half a million children in Africa and Asia go blind every year due to their diet being deficient in vitamin A, which is important for vision and the immune system. β-Carotene (CHEBI:17579), a red-orange pigment abundant in plants and fruit, is the primary dietary source of vitamin A worldwide. In the body, β-carotene is converted into vitamin A, which can exist in several forms (retinol, retinal, retinoic acid and retinyl esters). The role of vitamin A in the visual cycle is specifically related to the retinal form. Within the eye, 11-cis-retinal is bound to rhodopsin (rods) and iodopsin (cones) at conserved lysine residues. As light enters the eye, the 11-cis-retinal is isomerised to the all-trans form. This isomerisation induces a nervous signal along the optic nerve to the visual centre of the brain. The all-trans-form is subsequently recycled via a series of enzymatic reactions.A variety of sweet potato bred naturally to contain four to six times the amount of β-carotene than the average sweet potato has helped to stave off vitamin A deficiency in parts of Africa [1]. In a two year project headed by Christine Hotz at the International Food Policy Research Institute in Washington DC involving 10,000 households in Uganda, it was found that vitamin A intake doubled in women and young children who ate the biofortifed sweet potatoes compared with families that continued eating regular varieties [2]. Additionally, by the end of the study almost 90% of the children who ate the new strain had escaped vitamin A deficiency, compared with only 50% in a control group.
More controversial than the naturally bred sweet potatoes is Golden Rice, a variety of Oryza sativa rice produced via genetic engineering to be rich in β-carotene, of which ordinary white rice contains none. Critics of the modified rice, including Greenpeace, have claimed that the rice is impractical as people would need to consume huge amounts in order to obtain sufficient quantities of β-carotene. A recent study carried out by Guangwen Tang and co-workers at Tufts University in Boston, Massachusetts, has compared the vitamin A value of β-carotene in Golden Rice and in spinach with that of pure β-carotene in oil when consumed by children [3]. Analyses showed that the β-carotene in Golden Rice was as effective as pure β-carotene in oil and superior to that in spinach at providing vitamin A to children. A bowl of 100–150 grams of cooked Golden Rice (50 grams dry weight) can provide approximately 60% of the recommended daily intake of vitamin A. Although the results seem to counter the criticism, several obstacles will have to be overcome before Golden Rice becomes widely available.
Reference(s)
- http://www.newscientist.com/article/17-08-2012.
- Hotz, C., Loechl, C., Lubowa, A., Tumwine, J.K., Ndeezi, C., Masawi, A.N., Baingana, R., Carriquiry, A., de Brauw, A., Meenakshi, J.V. and Gilligan, D.O. (2012), Introduction of β-carotene-rich orange sweet potato in rural Uganda results in increased vitamin A intakes among children and women and improved vitamin A status among children. J. Nutr., doi:10.3945/jn.111.151829, published online 8 August 2012.
- Tang, G., Hu, Y., Yin S.-a., Wang, Y., Dallal, G.E., Grusak M.A. and Russell, R.M. (2012), β-Carotene in Golden Rice is as good as β-carotene in oil at providing vitamin A to children. Am. J. Clin. Nutr., 56, 658–664.
6th August 2012, thionitrous acid
Until the mid-1980s, nitric oxide (NO) – a short-lived, highly reactive, toxic gas – was best known as an air pollutant from internal combustion engines (but produced for and used on a huge scale in the Ostwald process for the manufacture of nitric acid). The independent discovery by Robert Furchgott, Louis Ignarro and Ferid Murad that nitric oxide was endogenously produced and acted as a signalling molecule between cells, playing a vital role in regulating blood pressure and circulation, was completely unexpected and opened up new areas of biomedical research. For their breakthrough, Professors Furchgott, Ignarro and Murad shared the Nobel Prize in Physiology or Medicine in 1998. During his Nobel acceptance speech, Furchgott noted the irony that Alfred Nobel, celebrated for his work with nitroglycerin and dynamite, had suffered from angina, for which he was prescribed nitroglycerin [1]. Furchgott, Ignarro and Murad's work led researchers to the explanation of why nitroglycerin is so effective in treating heart pain: it is actually a prodrug, being converted by mitochondrial aldehyde dehydrogenase (EC 1.2.1.3) in the body into nitric oxide, which is a natural vasodilator [2].More recently, hydrogen sulfide (H2S) – another reactive, toxic gas – has also been recognised as an important signalling molecule, with similar physiological effects to nitric oxide [3]. Now, work led by Milos R. Filipović of the Department of Chemistry and Pharmacy at the University of Erlangen-Nürnberg in Germany suggests that the effects of nitric oxide and hydrogen sulfide may be linked by thionitrous acid, HSNO (CHEBI:65308) [4], a molecule so reactive it has previously only been isolated and spectroscopically identified in an argon matrix at –261°C [5].
One important signalling pathway for nitric oxide involves S-nitrosation of the thiol groups of cysteine residues in proteins, affording S-nitrosated proteins. To date, over 3000 S-nitrosoproteins have been reported [6], and S-nitrosation is being seen to be as important as phosphorylation. Filipović and co-workers have shown that S-nitrosated proteins can react with hydrogen sulfide to produce thionitrous acid, which can be metabolised to produce NO(+), NO, and NO(–) species, each of which evokes distinct physiological responses. In addition, the group have shown that thionitrous acid can freely diffuse through membranes, so facilitating the transfer of nitroso groups from one protein to another in signalling pathways. Not only does their study explain some of the physiological effects ascribed to hydrogen sulfide, it suggests that thionitrous acid may play a key role in cellular redox regulation.
The background image is a Creative Commons licensed picture showing the production and diffusion of nitric oxide (NO) (white) in the cytoplasm (green) of clusters of conifer cells one hour after mechanical agitation [7]. NO diffuses into the culture medium around cell clusters. The large dark green circle inside each cell is the nucleus. Some nuclei may contain a nucleolus (light green). The small and irregular dark areas outside the nucleus and in the cytoplasm represent the vacuoles (laser confocal photomicrograph x400). NO was visualised with a fluorescent probe DAF-2 DA (diaminofluorescein diacetate). NO is derived from L-arginine and oxygen by nitric oxide synthase (NOS, EC 1.14.13.39) activity in conifers and humans.
Reference(s)
- http://www.nobelprize.org/nobel_prizes/medicine/laureates/1998/furchgott-speech_en.html
- Chen, Z., Foster, M.W., Zhang, J., Mao, L., Rockman, H.A., Kawamoto, T., Kitagawa, K., Nakayama, K.I., Hess, D.T. and Stamler, J.S. (2005), An essential role for mitochondrial aldehyde dehydrogenase in nitroglycerin bioactivation. Proc. Natl. Acad. Sci. U. S. A., 102(34), 12159–12164.
- Wagner, C.A. (2009), Hydrogen sulfide: a new gaseous signal molecule and blood pressure regulator. J. Nephrol., 22(2), 173–176.
- Filipovic, M.R., Miljkovic, J.Lj., Nauser, T., Royzen, M., Klos, K., Shubina, T., Koppenol, W.H., Lippard, S.J. and Ivanović-Burmazović, I. (2102), Chemical characterization of the smallest S-nitrosothiol, HSNO; cellular cross-talk of H2S and S-nitrosothiols. J. Am. Chem. Soc., 134(29), 12016–12027.
- Nonella, M., Huber, J.R. and Ha, T.-K. (1987), Photolytic preparation and isomerization of thionyl imide, thiocyanic acid, thionitrous acid, and nitrogen hydroxide sulfide in an argon matrix: an experimental and theoretical study. J. Phys. Chem., 91(20), 5203–5209.
- Hess, D.T. and Stamler, J.T. (2012), Regulation by S-nitrosylation of protein post-translational modification. J. Biol. Chem., 287(7), 4411–4418.
- Durzna, D.D. (2009), Arginine, scurvy and Cartier's "tree of life". J. Ethnobiol. Ethnomed., 5:5.
2nd July 2012, L-phenylalanine
L-Phenylalanine (CHEBI:17295; more commonly know just as 'phenylalanine' and abbreviated as Phe or F) is one of the twenty common DNA-encoded amino acids used in protein formation. It is also a precursor for L-tyrosine which is in turn converted into the signalling molecules dopamine, (R)-noradrenaline and (R)-adrenaline.The inability of an individual to metabolise L-phenylalanine is one of the characteristics of the genetic disorder phenylketonuria (PKU). Individuals who suffer from PKU have a mutation in the gene for the hepatic enzyme phenylalanine 4-monooxygenase (phenylalanine hydroxylase, PAH, EC 1.14.16.1) and must carefully regulate their intake of L-phenylalanine in order to avoid high levels accumulating in the blood as their bodies break down proteins into their component amino acids. PKU has all the characteristics of amyloid diseases, disorders characterised by inappropriate folding and aggregation of proteins and peptides to form toxic protein fibrils. Such disorders include the age-related neurodegenerative diseases Alzheimer's and Parkinson's. Aromatic amino acids such as L-phenylalanine are known to be important for accelerating the amyloid assembly process and, especially when the amyloid beta protein (Aβ) contains two adjacent L-phenylalanine residues, these seem to mediate the intermolecular reactions between polypeptide chains needed for fibril assembly as seen in Alzheimer's disease.
Now, in a recent publication in Nature Chemical Biology [1] a team from Tel-Aviv University led by Ehud Gazit, in collaboration with researchers from the University of Zurich, report a direct connection between PKU and Alzheimer's disease by demonstrating that L-phenylalanine can by itself form toxic fibrils. They found that at concentrations that exist in PKU, L-phenylalanine self-assemblies into amyloid-like fibrils. Gazit's team worked with mice genetically modified to have a PKU-like condition. These expressed antibodies against L-phenylalanine fibrils which were not found in non-diseased mice. Use of fluorescent antibody probes allowed unambiguous detection of the L-phenylalanine fibrils.
Gazit's results provide a greater understanding of the molecular nature of PKU amyloid diseases, and suggest that antibody-led approaches currently being developed to treat Alzheimer's and Parkinson's by targeting fibrils should work also with PKU. The team is currently seeking a partner in the pharma industry to help in their further investigations.
The background image is of Alzheimer's beta-amyloid fibrils, based on PDB ID 2BEG and rendered with the open-source PyMol visualisation system.
Reference(s)
- Adler-Abramovich, L., Vaks, L., Carny, O., Trudler, D., Magno, A., Caflisch, A., Frenkel, D. and Gazit, E. (2012) Phenylalanine assembly into toxic fibrils suggests amyloid etiology in phenylketonuria. Nature Chemical Biology advance online publication, 17 June 2012 (doi:10.1038/nchembio.1002).
1st June 2012, auranofin
Amoebiasis (previously known as entamoebiasis) is an infection of the intestines caused by ingestion of the amoeba Entamoeba histolytica. The infection is spread through ingestion of the cyst-form of the parasite, generally through ingestion of food or water contaminated with human faecal matter. Amoebiasis is therefore often endemic in areas of the world lacking modern sanitation systems.An estimated 90% of the 40 to 50 million people in the world infected by E. histolytica do not show any symptoms; the amoeba simply eats and digests bacteria and food particles in the intestine, which is protected from attack by a layer of mucus. However, as the Latin name histolytica suggests (histo-lytic = tissue destroying), the amoeba can be far from benign. If it does come into contact with the cells lining the intestine, it secretes the same substances it uses to digest bacteria, so destroying the cell membranes and resulting in penetration and digestion of human tissues. Symptoms range from fatigue, abdominal cramps and watery or bloody diarrhoea through to fever and vomiting. The insidious onset of symptoms (typically two to four weeks after initial infection) and their variability make diagnosis particularly difficult [1]. Once amoebae enter the bloodstream, they may be carried to other organs in the body, particularly the liver, causing liver abscess. In 1998, the World Health Organisation estimated that amoebiasis resulted in about 70,000 deaths annually, making it the fourth leading cause of death and the third leading cause of morbidity due to protozoan infections worldwide [2].
Non-invasive amoebiasis is commonly treated using paromomycin, but this is poorly absorbed from the gastrointestinal tract (most of the dose is eliminated unchanged in the faeces) so another antiprotozoal, metronidazole, is used to treat invasive amoebiasis. However, metronidazole has adverse effects [3], while potential resistance of E. histolytica to the drug is a growing problem [4,5].
Now, a collaboration of scientists from the University of California (UC), San Francisco, UC San Diego, Wake Forest School of Medicine at Winston-Salem, North Carolina, the University of Utah, Salt Lake City and the Centre for Research and Advanced Studies of the National Polytechnic Institute in Mexico City has developed an automated high-throughput screen to test a 910-member library consisting of both FDA-approved and unapproved bioactive compounds for activity against E. histolytica [6]. This involved overcoming the problems that E. histolytica is an anaerobe and that no rapid readout assay was available. The group found that auranofin (CHEBI:2922), a drug approved by the FDA 25 years ago for the treatment of rheumatoid arthritis, was ten times more potent against E. histolytica than metronidazole. In a mouse model of amoebic colitis and a hamster model of amoebic liver abscess, auranofin markedly decreased the number of parasites, damage from inflammation, and the size of liver abscesses.
The fact that auranofin has already been approved by the FDA for use in humans means that years of expensive development can be saved. The U.S. National Institutes of Health has granted auranofin "orphan-drug" status (used to identify a significant, newly developed or recognized treatment for a disease which affects fewer than 200,000 persons in the U.S.) and the UC San Diego hopes to conduct clinical trials in the near future.
The background image shows Trophozoites (the active, feeding stage) of Entamoeba histolytica with ingested erythrocytes.
Reference(s)
- Haque, R., Huston, C.D., Hughes, C.D., Houpt, E. and Petri, W.A., Jr. (2003), Amebiasis. N. Engl. J. Med., 348(16), 1565–1573.
- The world health report 1998 - Life in the 21st century: A vision for all. (World Health Organisation, Geneva, 1998).
- Krogstad, D.J. and Cedeno, J.R. (1988), Problems with current therapeutic regimens. In Ambiasis: Human Infection by Entamoeba histolytica (ed. Ravdin, J.I.) 741–748 (John Wiley & Sons, New York).
- Samarawickrema, N.A., Brown, D.M., Upcroft, J.A., Thammapalerd, N. and Upcroft, P. (1997), Involvement of superoxide dismutase and pyruvate:ferredoxin oxidoreductase in mechanisms of metronidazole resistance in Entamoeba histolytica. J. Antimicrob. Chemother., 40(6), 833–840.
- Wassmann, C., Hellberg, A., Tannich, E., and Bruchhaus, I. (1999), Metronidazole resistance in the protozoan parasite Entamoeba histolytica is associated with increased expression of iron-containing superoxide dismutase and peroxiredoxin and decreased expression of ferredoxin 1 and flavin reductase. J. Biol. Chem., 274(37), 26051–26056.
- Debnath, A., Parsonage, D., Andrade, R.M., He, C., Cobo, E.R., Hirata, K., Chen, S., García-Rivera, G., Orozco, E., Martínez, M.B., Gunatilleke, S.S., Barrios, A.M., Arkin, M.R., Poole, L.B., McKerrow, J.H., and Reed, S.L. (2012), A high-throughput drug screen for Entamoeba histolytica identifies a new lead and target. Nature Medicine, doi:10.1038/nm.2758, published online 30 May 2012.
8th May 2012, Imidacloprid
The recent decline in bee population, known as colony collapse disorder, is causing great concern amongst scientists because of the valuable ecosystem service they provide [1]. Two recent studies have indicated that a group of widely used pesticides may be partially to blame for declining bee populations. Neonicotinoids are a class of insecticides chemically related to nicotine and emerged in the mid-1990s as a relatively less toxic alternative to human-damaging pesticides. They are used to protect major crops and have become the world's most commonly used group of pesticides. However, exposure to sub-lethal amounts can also cause harm to non-target organisms, such as bees.In a study by David Goulson and co-workers at the University of Stirling, UK, colonies of the bumble bee Bombus terrestris were exposed in the laboratory to sub-lethal amounts of the neonicotinoid imidacloprid (CHEBI:5870) and subsequently allowed to develop naturally under field conditions [2]. The pesticide-dosed colonies were found to have gained significantly less weight and produced 85% fewer new queens compared with control colonies. A separate study headed by Mickaël Henry at the National Institute for Agricultural Research in Avignon, France, offers a possible explanation for these findings [3]. They fed low levels of another neonicotinoid, thiamethoxam, to European honey bees Apis mellifera and tagged the bees with radio-frequency identification microchips to monitor their movements. The researchers found that the dosed bees were much more likely to die away from their hives, and suggested that the pesticide caused impairment of the bees' navigation abilities so they could not find their way home.
While the findings don't conclusively explain global bee declines they are expected to add weight to the call for the use of neonicotinoids to be banned or regulated. Germany, France and Slovenia already have strict limits on their use, and US beekeepers recently petitioned the Environmental Protection Agency to ban another neonicotinoid, clothianidin. Regulatory agencies are considering new guidelines for risk assessment of pesticides to bees and conducting tests in conditions that mimic nature as close as possible. Currently, tests are typically done in the laboratory and only focus on whether the pesticides kill the bees, but do not detect other subtle effects that can prove critical in the wild.
Reference(s)
- Stokstad, E. (2012), Field research on bees raises concern about low-dose pesticides. Science, 335, 1155–1156..
- Whitehorn, P.R., O'Connor, S., Wackers, F.L. and Goulson D. (2012), Neonicotinoid pesticide reduces bumble bee colony growth and queen production. Science, 336, 351–352.
- Henry, M., Béguin, M., Requier, F., Rollin, O., Odoux, J.-F., Aupinel, P., Aptel, S., Tchamitchian, S. and Decourtye, A., (2012), A common pesticide decreases foraging success and survival in honey bees. Science, 336, 348–350.
2nd April 2012, 4-methylimidazole
4-Methylimidazole (CHEBI:40035) is produced commercially by cyclocondensation of formaldehyde and ammonia with methylglyoxal. It is used as a chemical intermediate, starting material, or component in the manufacture of pharmaceuticals, photographic chemicals, dyes and pigments, cleaning and agricultural chemicals, and rubber. It has been identified as a by-product of fermentation and from the browning of certain foods through the Maillard reaction between carbohydrates and organic amino-containing compounds. In particular, it is found in types of caramel colouring produced using ammonia-based processes. It has also been detected in mainstream and sidestream tobacco smoke. Because of its high potential for human exposure, a decade ago 4-methylimidazole was nominated by the US National Cancer Institute for a long-term study.The results of the two-year feed studies that were carried out, published in a 278-page report in 2007 [1] and subsequently in a 2008 paper [2], concluded that although there was no evidence of carcinogenic activity of 4-methylimidazole in male rats, there was equivocal evidence of such activity in female rats based on increased incidences of mononuclear cell leukaemia. There was also clear evidence of carcinogenic activity in both male and female mice based on increased incidences of alveolar/bronchiolar neoplasms.
It was largely on the basis of this report that in January 2011 the State of California added 4-methylimidazole to its list of probable carcinogens [3] and stipulated the "No Significant Risk Level" intake to be 16 μg per day, an amount considerably less than that ingested by regular drinkers of the carbonated beverage cola. Hence on March 8th 2012 both the two main producers of cola, Coca-Cola and PepsiCo, announced [4] that they had lowered levels of 4-methylimidazole in the caramel colouring used in the manufacture of their drinks so as to avoid having to label their products with a cancer warning to comply with Californian law. However, both manufacturers insist that the drinks pose no health threats to humans and that, with the exception of the reduction in 4-methylimidazole content for the US market, recipes will not change. Indeed, the recipes are not changing in Europe at all, as there are no similar restrictions there on acceptable levels of 4-methylimidazole.
Reference(s)
- National Toxicology Program (2007) Toxicology and Carcinogenesis Studies of 4-Methylimidazole (CAS No. 822-36-6) in F344/N Rats and B6C3F1 Mice (Feed Studies). Technical Report Series No. 535. NIH Publication No.05-4471. U.S. Department of Health and Human Services, Public Health Service, National Institutes of Health; Research Triangle Park, NC.
- Chan P.C., Hill G.D., Kissling G.E. and Nyska A. (2008) Toxicity and carcinogenicity studies of 4-methylimidazole in F344/N rats and B6C3F1 mice. Arch. Toxicol., 82, 45–53.
- California Office of Environmental Health Hazard Assessment (2011) Proposition 65. Chemical listed effective January 7, 2011 as known to the State of California to cause cancer: 4-methylimidazole.
- The Coca-Cola Company, Coca-Cola Statement Regarding Caramel in our Beverages, press release, March 9, 2012.
5th March 2012, Ethanol
In one of the more unusual quirks of nature, an insect species has been found to use alcohol (ethanol, CHEBI:16236) for self-medication to ward off a blood-borne parasite [1]. Fruit flies of the species Drosophila melanogaster have developed an extremely high tolerance to alcohol over evolutionary time owing to their living in and feeding on rotting and fermented fruit. A recent study carried out by a research team headed by Todd Schlenke at Emory University (Atlanta, Georgia) has shown that the flies actually seek out and utilise the alcohol to help prevent predation from tiny parasitic wasps, which are their major killers [2]. The wasp mothers deposit their eggs on the inside of fruit fly larvae along with a venom to suppress their hosts' immune response. The hatchlings then eat the young flies from the inside out, eventually emerging as adult wasps from the remains of the fruit fly pupae. Fortunately for the flies, the high alcohol concentration in their bodies is harmless to them but deadly to the wasps.Schlenke and his team released healthy and wasp-infected fruit fly larvae into Petri dishes that contained both regular food and food laced with alcohol, allowing them to freely move to either side. After 24 hours, 80% of the infected larvae were on the alcohol side of the dish, compared with only 30% of the non-infected ones. With infected fruit flies that had consumed alcohol-laced food, the wasps were killed in approximately 60% of the cases, but grew normally in all flies that fed on regular food. Furthermore, the wasps were found to be discouraged from laying their eggs into alcohol-soaked larvae. The team repeated the experiment using the species of wasp (Leptopilina boulardi) that lays its eggs specifically in D. melanogaster rather than the generalist wasp (L. heterotoma). Here, the alcohol diet was less effective, killing the specialist wasps in only 10% of the cases. These results indicate that the specialist wasp has also been able to adapt to the alcohol-fused habitat of D. melanogaster. The researchers hope that their data will lead to further studies of the protective effects of alcohol consumption in other organisms, including humans.
The background image shows a specimen of the fruit fly Drosophila melanogaster.
Reference(s)
- http://www.newscientist.com/article/dn21493-parasiteplagued-flies-selfmedicate-on-booze.html
- Milan, N.F., Kacsoh, B.Z. and Schlenke, T.A. (2012), Alcohol consumption as self-medication against blood-borne parasites in the fruit fly. Current Biology, doi:10.1016/j.cub.2012.01.045, published online 16 February 2012.
6th February 2012, (+)-artemisinin
There are five species of Plasmodium protazoan parasites which cause malaria in humans, of which Plasmodium falciparum is the deadliest [1,2], being responsible for almost all of the estimated 655,000 deaths from malaria in 2010 [3], the majority being of young children. Around 90% of the malaria-related deaths occur in sub-Saharan Africa, where P. falciparum is the cause of more than 75% of malarial infections.As part of a secret anti-malarial drug discovery project set up by the Chinese army in 1967, Tu Youyou, a medical scientist at the Academy of Chinese Medicine (now the China Academy of Chinese Medical Research), in Beijing, screened over 2,000 recipes for traditional Chinese medicines. By 1971, her team had prepared 380 extracts from 200 herbs. They then assessed whether the substances could clear Plasmodia from the bloodstream of infected mice. One of the preparations she tested, described in a fourth century book titled Handbook of Prescriptions for Emergencies, used leaves of Artemisia annua (sweet wormwood, a common herb found in many parts of the world). Initially it was ineffective, as it had been extracted with boiling water which had destroyed the active ingredient. By extracting at lower temperatures using ether as the solvent, however, the preparation completely cleared Plasmodium from the blood in both mice and monkeys [4]. By the end of 1972, she and her team had isolated the active ingredient. This substance, named qinghaosu by the Chinese, remained unknown in the west until 1979 [5]. Artemisinin (CHEBI:223316), as the drug is best known in the west, has since saved millions of lives. It is the most effective treatment currently available for multi-drug-resistant forms of malaria, and artemisinin combination therapies (ACTs) are recommended as first-line drugs by the World Health Organisation [3]. Tu Youyou was awarded the prestigious Lasker-DeBakey Clinical Medical Research Award for her discovery in 2011.
Although a total synthesis of artemisinin has been described [6], it is not considered to be a viable route for satisfying the demand for artemisinin, which is now one of the few high-volume drugs to be entirely produced from a plant-based source. As Artemisia annua is an annual, supplies of artemisinin are seasonal and the price fluctuates widely. ACT drugs are consequently more expensive than other malaria treatments, even though Novartis and Sanofi provide the former on a non-profit basis [7]. By using artemisinic acid, a much less complex, bicyclic precursor as a starting point for synthesis, however, artemisinin could soon become cheaper and more readily available. Artemisinic acid is also obtainable from Artemisia annua, though in higher yields than artemisinin. Better still, artemisinic acid can be produced by engineered yeast, using a process invented by a group led by Jay D. Keasling at the University of California, Berkeley [8] and developed commercially by Amyris. Amyris have licensed their technology to Sanofi, who aim to bring artemisinin-based drugs to the market by 2013 [9]. The large-scale transformation of artemisinic acid to artemisinin is still a massive challenge, however, requiring the creation of three of the artemisinin's rings and the generation of the endoperoxide group that is crucial to the antimalarial activity.
Thanks to a recent publication by François Levésque and Peter H. Seeberger at the Max-Planck Institute for Colloids and Interfaces in Germany [10], however, the task could have been made considerably easier. They describe the combination of three separate steps in the transformation into a single continuous flow process. Of particular note is an oxidation step requiring the photochemical generation of singlet dioxygen. Reactions involving this highly reactive species are very difficult to perform on a large scale in conventional batch systems, due to the low rate of mass transfer of oxygen gas into the solution and the formation of potentially explosive hydroperoxides. However, Seeberger and Levésque have shown that they can generate singlet dioxygen on a preparative scale by simply wrapping the tubing containing the reactant flow around a cooled mercury lamp [11]. Their setup can now produce around 800 g of artemisin per day, meaning that the entire world demand for the drug, estimated at roughly 225 million doses, could be met by just 400 such reactors [9].
The background image shows an Anopheles gambiae mosquito feeding.
Reference(s)
- Perlmann, P. and Troye-Blomberg, M. (2000), Malaria blood-stage infection and its control by the immune system. Folia Biol. (Prague, Czech. Repub.), 46(6), 210–218.
- Snow, R.W., Guerra, C.A., Noor, A.M., Myint, H.Y. and Hay, S.I. (2005), The global distribution of clinical episodes of Plasmodium falciparum malaria. Nature, 434, 214–217.
- World Malaria Report 2011.(World Health Organisation, Geneva, 2011).
- Miller, L.H. and Su, X. (2011), Artemisinin: discovery from the Chinese herbal garden. Cell, 146(6), 855–858.
- Qinghaosu Antimalarial Coordinating Research Group (1979), Chin. Med. J., 12, 811–816.
- Schmid, G. and Hofheinz, W. (1983), J. Am. Chem. Soc.,, 105, 625–627.
- Artemisinin combination therapies,(The CNAP Artemisia Research Project, University of York).
- Ro, D.-K., Paradise, E.M., Ouellet, M., Fisher, K.J., Newman, K.L., Ndungu, J.M., Ho, K.A., Eachus, R.A., Ham, T.S., Kirby, J., Chang, M.C.Y., Withers, S.T., Shiba, Y., Sarpong, R., and Keasling, J.D. (2006), Production of the antimalarial drug precursor artemisinic acid in engineered yeast. Nature, 440, 940–943.
- Halford, B. (2012), Artemisinin goes with the flow. Chem. Eng. News, 90(4), 4.
- Levésque, F. and Seeberger, P.H. (2012), Continuous-flow synthesis of the anti-malaria drug artemisinin. Angew. Chem., Int. Ed., 51, in press.
- Levésque, F. and Seeberger, P.H. (2011), Highly efficient continuous flow reactions using singlet oxygen as a “green” reagent. Org. Lett., 13, 5008–5011.

9th January 2012, Mercury
Mercury (CHEBI:16170) is the only metallic element that is a liquid at standard temperature and pressure. Along with most of its compounds it is extremely toxic, causing both chronic and acute poisoning. Moreover, it has a tendency to accumulate in organisms and increase in concentration up through the food chain, and therefore monitoring of its levels in the environment is vitally important.For this reason Aurore Aubail, working at the National Environmental Research Institute of Denmark, together with colleagues from France and Norway, have studied the temporal changes in mercury pollution exposure over the period 1964 to 2003 using polar bear teeth as a measure of the mercury exposure, and they now report their findings in the latest issue of Journal of Environmental Monitoring [1]. Polar bears sit on top of the Arctic food chain and are therefore particularly susceptible to accumulating high levels of mercury within tissues. Their teeth are regarded as an especially good matrix of the mercury exposure as, unlike soft tissue, they are not remodelled throughout the animal’s life.
Teeth from 87 polar bear skulls were therefore analysed using solid sample atomic absorption spectrophotometry. In addition the relative abundances of carbon (13C/12C) and nitrogen (15N/14N) stable isotopes were measured using isotope-ratio mass spectrometry to provide information about any changes in feeding habits or habitats. The results showed (i) a significant decrease in mercury concentrations in the dental tissue over the four decades and (ii) that there was no significant temporal trend in the δ15N or δ 13C signatures, thus eliminating variations in feeding habits or habitats. Aubail and her co-workers suggest that this decreasing temporal trend for mercury levels in the dental tissue may result from an overall decrease in mercury emissions from Europe and North America. When interviewed for Chemistry World, Aubail expressed the hope that the use of polar bear teeth to study mercury would continue as one of the methods of studying long-term trends of pollutants [2].
Reference(s)
- Aubail, A., Dietz, R, Rigét, F., Sonne, C., Wiig, Ø and Caurant, F. (2012), Temporal trend of mercury in polar bears (Ursus maritimus) from Svalbard using teeth as a biomonitoring tissue. J. Environment. Monit., 14, 56–63.
- Arctic biting back over mercury pollution, Chemistry World, 9 December 2011.
2011
5th December 2011, Pyrrolidin-2-one
Although spiders seem safe in their webs, in some parts of the world they are vulnerable to attack from armies of marauding ants. Surprisingly, ants are rarely found on the webs spun by batik golden web spiders (Nephila antipodiana), which live alongside many predatory ant species in south-east Asia [1]. Mark Elgar at the University of Melbourne, Australia, and Daiqin Li at the National University of Singapore analysed the webs of 21 batik golden web spiders and found that their silk contained pyrrolidin-2-one (CHEBI:36592). After stripping strands of the silk of chemicals, ants were lured with bait across a bridge constructed from the stripped thread. After reapplying pyrrolidin-2-one to the stripped thread, the ants were then deterred from following a trail of food back over it [2].Furthermore, the chemical is only produced by adult and large juvenile spiders, while small and immature spiders, whose threads are sufficiently thin to make them inaccessible to ants, do not produce any. This suggests that production of pyrrolidin-2-one is actually a chemical response to the threat of predation, rather than a simple by-product of silk synthesis. These findings could pave the way for development of new insect repellents for humans.
Other current applications of pyrrolidin-2-one include use in industry as a high boiling non-corrosive polar solvent and as an intermediate in the manufacture of polymeric compounds, such as poly(vinylpyrrolidone).
Reference(s)
- http://www.newscientist.com/article/mg21228405.600-spider-webs-protected-by-chemical-ants-hate.html
- Zhang, S., Koh, T.H., Seah, W.K., Lai, Y.H., Elgar, M.A. and Li D. (2011), A novel property of spider silk: chemical defence against ants. Proc. R. Soc. B, doi:10.1098/rspb.2011.2193, published online 23 November 2011.
7th November 2011, Acetylsalicylic acid
The contributions of Dr. Thomas John MacLagan (1838–1903) to medicine were at one time ranked as equivalent to those of Joseph Lister and James Young Simpson, but have been largely forgotten [1]. MacLagan was a medical superintendent at Dundee Royal Infirmary from 1864 to 1866, during which time he had to cope with a major fever epidemic and became noted for pioneering the clinical use of thermometers. While working as a medical practitioner in Dundee (1869–1779), he carried out research into the use of salicin, a chemical extracted from willow bark. In 1874 he began using salicin for the treatment of rheumatism, and was the first to use salicin to cure acute articular rheumatism. His work was taken up by researchers in Germany, and in 1897 chemists at Bayer AG produced a synthetically altered version of salicin, acetylsalicylic acid (CHEBI:15365) (which had actually been prepared over forty years earlier by a French chemist, Charles Frédéric Gerhardt [2], though in a rather impure form). Bayer AG sold acetylsalicylic acid around the world under the trade name by which it is now best known, Aspirin. Although its use declined following the introduction of more effective alternatives (e.g. paracetamol, also known as acetaminophen, in 1956; ibuprofen in 1969), it rose again in the 1990s when it was shown to be effective as an anticoagulant agent and so was used as a preventative treatment for heart attack and stroke. In 2002, worldwide use was estimated as 40,000 tonnes – equivalent to around 120 billion tablets – per year [3].More recently, it has become apparent that aspirin may find a potential new use as an agent for the prevention of colorectal cancer. Also known as bowel cancer, this is the second most common cancer in developed countries, with about 1 million new cases and over 600,000 deaths worldwide each year. Late last year, a 20-year follow-up of five pooled randomised trials of cardiovascular disease prevention assessing the effect of aspirin on incidence and mortality of colorectal cancer suggested that a daily dose of at least 75 mg of aspirin taken for several years reduced the 20-year risk of the cancer by 24%, while associated mortality was reduced by 35% [4]. However, a final conclusion on the ability of aspirin to prevent colorectal cancer still required a randomised trial of aspirin with colorectal cancer as the primary target. Now, the results of a study involving 861 carriers of the most common inherited colorectal cancer, Lynch syndrome (often called hereditary nonpolyposis colorectal cancer or HNPCC), have now been described in a major report by Sir John Burn and colleagues [5] which has generated international news coverage. Their study showed that carriers who took a daily dose of 600 mg of aspirin per day for an average of 25 months suffered significantly fewer cancers than those in the control group. Combined, these results and previous evidence suggest that aspirin should be seriously considered for the prevention of colorectal cancer, particularly in those who are at high risk of developing the disease.
The background image shows willows, a source of salicin, on the River Avon, east of Stratford-upon-Avon.
Reference(s)
- Stewart, W.K. and Fleming, L.W. (1987) Perthshire pioneer of anti-inflammatory agents (Thomas John Maclagan). Scott. Med. J., 32(5), 141–146. PMID: 3327165.
- Gerhardt, C. (1853) Untersuchungen über die wasserfreien organischen Säuren. Ann. Chem., 87(2), 149–179.
- Warner, T.D. and Mitchell, J.A. (2002) Cyclooxygenase-3 (COX-3): Filling in the gaps toward a COX continuum? PNAS, 99(21), 13371–3373.
- Rothwell, P.M., Wilson, M., Elwin, C.-E., Norrving, B., Algra, A., Warlow, C.P. and Meade T.W. (2010) Long-term effect of aspirin on colorectal cancer incidence and mortality: 20-year follow-up of five randomised trials. Lancet, 376, 1741–1750.
- Burn, J., Gerdes, A.-M., Macrae, F., Mecklin, J.-P., Moeslein, G., Olschwang, S., Eccles, D., Evans, D.G., Maher, E.R., Bertario, L., Bisgaard, M.-L., Dunlop, M.G., Ho, J.W.C., Hodgson, S.V., Lindblom, A., Lubinski, J. Morrison, P.J., Murday, V., Ramesar, R., Side, L., Scott, R.J., Thomas, H.J.W., Vasen, H.F., Barker, G., Crawford, G., Elliott, F., Movahedi, M., Pylvanainen, K., Wijnen, J.T., Fodde, R., Lynch, H.T., Mathers, J.C. and Bishop, D.T. (2011) Long-term effect of aspirin on cancer risk in carriers of hereditary colorectal cancer: an analysis from the CAPP2 randomised controlled trial. Lancet, doi:10.1016/S0140-6736(11)61049-0, published online 28 October 2011.
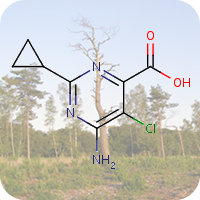
3rd October 2011, Aminocyclopyrachlor
Aminocyclopyrachlor (CHEBI:62952) was developed as a selective low-toxicity herbicide by the major American chemical company DuPont and was marketed by them as its potassium salt under the trade name Imprelis. However, in August of this year [less than a year following approval by the US Environmental Protection Agency (EPA)], DuPont voluntarily began a recall programme following several thousands of complaints that the product had caused significant damage to trees, particularly Norway spruce (Pinus abies) and the various species of white pine (e.g. Pinus strobus) [1].Aminocyclopyrachlor is a synthetic auxin with a substituted pyrimidine structure. Although the mechanism of its action has not been fully established, one theory is that it works in a manner similar to that of other synthetic herbicides such as clopyralidand aminopyralid, namely by interfering with a plant's hormonal balance in order to override its ability to control cellular metabolism at the most active growth points (shoots and roots) [2]. DuPont had been marketing Imprelis to provide preemergent and/or postemergent control of broadleaf weeds on non-food use sites, such as rights of way, wildlife management areas and recreational areas (e.g. golf courses), as well as in commercial properties such as turf farms. It is possible that the method of application, involving treatment of the grassland at a time when adjacent conifers are growing and metabolising at high rates, may be one of the main causes for the detrimental effect on the conifers, effects principally characterised by such symptoms as browning of tree shoots and needles and stunting of fresh growth.
The EPA issued an order halting the sale of Imprelis two weeks after DuPont's voluntary withdrawal of the product [3].
Reference(s)
- E. I. du Pont de Nemours and Company, Imprelis Facts information sheet, August 2011.
- Trusty, S. (2011) Industry Advancements: What«s New in Weed Control. Sports Field Management, January 2011.
- U.S. Environmental Protection Agency, News release, EPA Issues Stop Sale Order to DuPont on Sale and Distribution of Imprelis Herbicide, August 11, 2011.

5th September 2011, Tetraethyllead
The recently deceased Emeritus Professor Derek Bryce-Smith had a long and distinguished career at the University of Reading, spanning a wide variety of interests including organometallic chemistry, radical chemistry and photochemistry. However, he will arguably be best remembered for alerting the world to the dangers of tetraethyllead (CHEBI:30182), which was used for several decades as an anti-knock additive in petrol. Ridiculed by the oil industry and marginalised for several years, he lived to see the eventual banning of leaded fuel in almost every country. "It was a very lonely battle for a very long time," he said. "A lot of my colleagues looked at me sideways, because many research chemists are in debt to the oil industry, which provides them with money for research."Tetraethyllead consists of a lead atom bonded to a tetrahedral arrangement of four ethyl groups. The C-Pb bond is relatively weak, and in the hot environment of an internal combustion engine it undergoes fission to produce lead, a potent and insidious neurotoxin, and ethyl radicals which can help terminate the combustion process by radical reactions and prevent "knocking" (uncontrolled pre-ignition of fuel causing a pinging noise in the engine, which reduces efficiency and leads to eventual damage) [1]. The lead formed on combustion is subsequently dispersed into the atmosphere via the exhaust. The discovery of the environmental and health damage caused by the lead, as well as the incompatibility of lead with catalytic converters, resulted in the phasing out of leaded fuel and development of replacement anti-knock additives, such as MTBE, ferrocene, toluene and isooctane.
Reference(s)
- Seyferth, D. (2003), The rise and fall of tetraethyllead. 2. Organometallics 22, 5154–5178.

1st August 2011, Roxarsone
The antiprotozoal (coccidiostat) properties of roxarsone (CHEBI:35817), an organoarsenic compound first synthesised over a hundred years ago [1], were responsible for it becoming, on March 21st 1944, the first arsenic-based product approved for use in animal feeds by the U.S. authorities. For over half a century, roxarsone (also known by its trade name, 3-NitroTM) has been used as a feed additive in the poultry industry to control coccidiosis (a parasitic disease that infects the intestinal tract of poultry) and promote growth, particularly in broiler chickens.In its original organic form, the arsenic in roxarsone is relatively benign, being non-carcinogenic and less toxic than the inorganic forms of arsenic, arsenite and arsenate, which are now classed as human carcinogens by the U.S. Environmental Protection Agency, and can cause immunological and neurological problems on long-term exposure [2]. Very little roxarsone is retained in the chicken meat (the U.S. Food and Drug Administration (FDA) limit is 0.5 ppm in chicken muscle tissue), and nearly all is excreted unchanged, meaning that in the U.S. alone, an estimated 1,000-2,000 tonnes of roxarsone and its degradation products have been added to the environment each year by the disposal of poultry litter, which is either spread on agricultural fields near the chicken houses, or made into fertilizer pellets for use on domestic lawns and gardens [3].
Recognition that roxarsone was ultimately converted by bacteria into inorganic (and hence water-soluble) forms of arsenic in the environment [4–7], resulted in increased public concern over the use of roxarsone, as well as law suits contending that arsenic in poultry litter had caused ill effects in humans. The European Union banned the use of arsenic in animal feeds in 1999 [8], while in the U.S., Tyson Foods, the country's largest poultry producer, stopped using arsenic compounds in 2004. Even so, roxarsone remained part of the diet for the majority of the 9 billion broiler chickens produced in the U.S. each year [3]. In a recent FDA study [9], scientists found that levels of inorganic arsenic in the livers of chickens treated with roxarsone were increased relative to levels in the livers of the untreated control chickens. Even though the levels found are not thought to pose any risk to health, on June 8, 2011 Alpharma, LLC, (now a part of Pfizer Animal Health), who are the U.S. manufacturers, announced that they would voluntarily suspend sales of roxarsone within 30 days, so giving animal producers time to move to other treatment strategies [10,11]. It now seems likely that the future of nitarsone (trade name HistostatTM, also manufactured by Alpharma), a compound closely related to roxarsone and now the last remaining organoarsenic feed additive, is in the balance.
The background image shows five-day-old Cornish-Rock (Cornish Cross) broiler chicks in a brooder.
Reference(s)
- Benda, D. and Bertheim, A. (1911) Über Nitro-oxy-aryl-arsinsäuren. Ber., 44, 3445–3448.
- http://www.epa.gov/ttn/atw/hlthef/arsenic.html.
- Hileman, B. (2007), Arsenic in chicken production. Chem. Eng. News, 85(15), 34–35.
- Garbarino, J.R., Bednar, A.J., Rutherford, D.W., Beyer, R.S. and Wershaw, R.L. (2003), Environmental fate of roxarsone in poultry litter. I. Degradation of roxarsone during composting. Environ. Sci. Technol., 37, 1509–1514.
- Rutherford, D.W.,Bednar, A.J., Garbarino, J.R., Needham, R., Staver, K.W.,Wershaw, R.L. (2003), Environmental fate of roxarsone in poultry litter. Part II. Mobility of arsenic in soils amended with poultry litter. Environ. Sci. Technol.,, 37, 1515–1520.
- Cortinas, I., Field, J.A., Kopplin, M., Garbarino, J.R., Gandolfi, A.J. and Sierra-Alvarez, R. (2006), Anaerobic biotransformation of roxarsone and related N-substituted phenylarsonic acids. Environ. Sci. Technol., 40, 2951–2957.
- Stolz, J.F., Perera, E., Kilonzo, B., Kail, B., Crable, B., Fisher, E., Ranganathan, M., Wormer, L. and Partha Basu, P. (2007), Biotransformation of 3-nitro-4-hydroxybenzene arsonic acid (roxarsone) and release of inorganic arsenic by Clostridium species. Environ. Sci. Technol., 41, 818–823.
- Commission Directive 2009/141/EC.
- FDA Study 275.30 (2011), Arsenic speciation in broiler chickens. a) Summary Final Report. b) Amendments to final report. c) Analyst's report. d) Statistician's Report.
- Pfizer To Suspend Sale of 3-Nitro (Roxarsone) in the United States (Alpharma news release), June 8, 2011.
- FDA: Pfizer will voluntarily suspend sale of animal drug 3-Nitro (FDA press release), June 8, 2011
4th July 2011, Bendroflumethiazide
One of the more recent controversies involving the use of performance-enhancing drugs in sport has involved Manchester City footballer Kolo Touré. In February 2011, the player tested positive for a specified substance, subsequently found to be the banned diuretic bendroflumethiazide (CHEBI:3013), which he blamed on the use of dietary water tablets belonging to his wife. Touré admitted the offence, which contravened Regulation 3 of the Football Association Doping Regulations 2010-11 and was suspended from all footballing activities for six months [1], escaping the recommended two-year ban because the Football Association's independent regulatory commission ruled his intent was not to mask drug use, but instead to control his weight [2].Bendroflumethiazide belongs to a group of drugs called thiazide diuretics. These act inside the kidney, removing water from the blood and turning it into urine, through increasing the removal of salts such as potassium and sodium salts from the blood. This removal of salts causes water to be drawn out of the blood and into the kidneys, at a faster rate. Thiazides are also used to treat hypertension, although the mechanism of action is not fully understood.
Reference(s)

6th June 2011, Electron
Knowing something as apparently simple as whether electrons are round or egg-shaped has far-reaching ramifications for testing some of the theories of particle physics and for the types of particles that could be detected at high-energy accelerators, such as the Large Hadron Collider. The Standard Model of particle physics predicts that the electron (CHEBI:10545) should behave as though it were almost perfectly spherical, with the distortion from a perfect sphere being far too small to detect. However, this cannot account for the observed predominance of matter over antimatter in the universe, and so has led physicists to propose extensions and refinements to the Standard Model, known as supersymmetry, in which the electron would have a much more distorted shape than the Standard Model suggests - so much so, that the distortion should be detectable.Recently, a team from Imperial College in London has reported on an experiment spanning more than a decade and resulting in the most accurate measurements of the shape of the electron yet [1]. Their findings suggest that the electron differs from being perfectly round by less than 1 x 10-27 cm, meaning that if the electron were magnified to the size of the solar system, it would still appear spherical to within the width of a human hair. Although the findings do not support the theory of supersymmetry, they do not rule it out. The researchers are now refining their methods to improve the precision of their measurements still further.
The image shows a beam of electrons moving in a circle (cyclotron motion) within a glass bulb.
Reference(s)
- Hudson, J.J., Kara, D.M., Smallman, I.J., Sauer, B.E., Tarbutt, M.R., and Hinds, E.A. (2011), Improved measurement of the shape of the electron. Nature, 473, 493–496.
4th May 2011, Choline
Choline (CHEBI:15354) is a water-soluble, semi-essential nutrient similar to the B-vitamins. Long recognised as an important promoter of good health, it is synthesised by the body, but dietary supplementation is required to ensure adequate intake. Sources of choline include soybeans, egg yolk, beef, cauliflower and navy beans. A lipotropic agent, choline is essential in the brain development of foetuses and infants [1]. It has also been shown to improve cognitive function in adults; to enhance alertness and mood; and to support liver and cardiovascular health [2]. It is used in the treatment of liver disorders, Alzheimer's Disease, bipolar disorder and atherosclerosis (fat build-up in the arteries). Its most commonly available form is lecithin, a phospholipid mixture containing 20-90% active ingredient phosphatidylcholine, but healthy amounts of choline and its metabolites trimethylamine N-oxide (TMAO) and glycine betaine [3] occur in fruits, and direct-to-consumer dietary supplements are freely available for human consumption, as well as for use in livestock feed.Cardiovascular disease is the world's largest killer, with many risk factors which interact in myriad ways. Recent ground-breaking work in metabolomics (the study of global metabolite profiles in an organism under a given set of conditions) sheds some light on how these factors may interact differentially to produce the variable susceptibilities to heart disease experienced by different individuals, even while on identical diets. In particular, the Hazen group at the Lerner Research Institute in Cleveland has reported its discovery of a pathway linking dietary lipid intake and intestinal flora with increased risk of heart disease [4,5,6]. Eighteen small molecules were found to be associated with atherosclerotic plaque build-up in the arteries, three of which - choline, TMAO and betaine - were especially good predictors of atherosclerosis. This finding represents a unique addition to cardiovascular disease pathogenesis in implicating not just the common dietary supplements choline and its metabolites, but also in identifying intestinal flora as obligatory participants in atherogenesis. The findings suggest that, since these molecules can serve as heart disease markers, a blood test for any of them may be able to identify those at greater risk for heart disease.
A caveat in the interpretation of these results: the mice in the Hazen study were of a breed already genetically prone to heart disease, and were fed huge amounts of choline, much more than would normally be consumed in day-to-day living. Nevertheless, the study is useful in raising the question of the need to consider risk vs. benefits of some commonly used supplements. It also offers the prospect of the potential design of healthy-promoting yoghurt that acts by changing the demographics of intestinal probiotics, or drugs to block the production of harmful metabolites.
In conclusion, metabolomics studies have identified, through the discovery of novel heart disease markers, potential tools for improved diagnosis, prevention and treatment of heart disease.
Reference(s)
- Sentongo, T.A., Kumar., P., Karza, K., Keys, L., Iyer, K. and Buchman, A.L. (2010), Whole-blood-free choline and choline metabolites in infants who require chronic parenteral nutrition therapy. J. Pediatr. Gastroenterol. Nutr. 50, 194-9.
- Ueland, P.M. (2010), Choline and betaine in health and disease. J. Inherit. Metab. Dis. 34, 3-15.
- Babb, S.M., Ke, Y., Lange, N., Kaufman, M.J., Renshaw, P.F. and Cohen, B.M. (2004), Oral choline increases choline metabolites in human brain. Psychiatry Res. 130, 1-9.
- Wang, Z., Klipfell, E., Bennett, B.J., Koeth, R., Levison, B.S., DuGar, B., Feldstein, A.E., Britt, E.B., Fu, X., Chung, Y.M., Wu, W., Schauer, P., Smith, J.D., Allayee, H., Tang, W.H.W., DiDonato., J.A., Lusis, A.J. and Hazen, S.L. (2011), Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 472, 57-63.
- Carr, E. (2011), A role for gut flora in cardiovascular disease: you are what they eat. Cambridge Medicine Journal.
- Lerner Research Institute (2011), Common dietary fat and intestinal microbes linked to heart disease. ScienceDaily.
4th April 2011, Amphotericin B
Amphotericin B (ChEBI:2682) is a highly potent antifungal drug, which has been in widespread clinical use since the 1960's [1]. Its mode of action involves formation of molecular pores on fungal cell membranes. These transmembrane channels allow ions to leak from the cell, resulting in its death. The presence of a sterol, such as cholesterol or ergosterol, in the cell is required for formation of the ion channels. However, it is the exact nature and extent of the interaction between the fungicide and sterols in the cell that has hitherto baffled scientists for more than 50 years [2].Recent work carried out by a team of scientists from the University of Illinois at Urbana-Champaign led by Martin Burke have solved this mystery by performing a highly sensitive isothermal titration calorimetry assay [3], which has proved that direct binding between fungicide and sterol definitely occurs. By using intricate synthetic techniques, the researchers prepared several derivatives of amphotericin lacking the carboxylic acid and sugar functional groups [3], which were thought to be involved in sterol binding. By repeating the isothermal titration calorimetry assay with the synthetic derivatives, it was proved unequivocally that the sugar group alone is required for sterol binding to form ion channels and destroy fungal cells.
The enhanced understanding of the mode of action has potential to reduce the toxic side-effects of amphotericin B, which include chills, fever, hypotension, nausea and headaches. In addition, there is also the possibility to develop new drugs to replace faulty proteins in human cells and to correct ion-channel dysfunction, which is responsible for several diseases, including cystic fibrosis.
Reference(s)
- Sanglard, D. and Odds, F.C. (2002), Resistance of Candida species to antifungal agents: molecular mechanisms and clinical consequences. Lancet Infect. Dis. 2, 666–669.
- http://www.rsc.org/chemistryworld/News/2011/March/09031102.asp.
- Palacios, D.S, Dailey, I., Siebert, D.M., Wilcock, B.C. and Burke M.D. (2011), Synthesis-enabled functional group deletions reveal key underpinnings of amphotericin B ion channel and antifungal activities. Proc. Natl. Acad. Sci. U.S.A. doi:10.1073/pnas.1015023108.
9 March 2011, Thiopental sodium
Over 75 years has elapsed since thiopental sodium (CHEBI:9561) was first used clinically as an intravenous anaesthetic. Also called sodium thiopental and more commonly known in the UK as sodium thiopentone, it is a barbiturate, so acts on the GABAA receptors to cause a marked decrease in neuronal activity. For this reason, overdoses can be fatal, and were initially an important factor in mortality associated with the use of the drug, particularly for trauma patients who were in a state of shock. For many years, it was widely believed that at the Japanese attack on the American bases in Hawaii in December 1941, ..."i.v. anaesthesia was the cause of more fatalities among the servicemen at Pearl Harbor than were the enemy bombs" [1]. Only decades later, with the release of hospital records through U.S. freedom of information legislation, did it become clear that the rumoured death rate was greatly exaggerated. For example, of 344 wounded admitted to Tripler Army Hospital during the morning of December 7th 1941, only 13 did not survive [2]. With the publication of improved dosing procedures for trauma cases in January 1943 [3], thiopental sodium became an invaluable drug for the rapid induction (30 seconds) of general anaesthesia in both humans and animals. (It is not used to maintain anaesthesia due to prolonged recovery times; this is normally done with an inhaled anaesthetic agent.) It is still used to provide general anaesthesia hundreds of thousands of times each year in the U.K. alone [4].Thiopental sodium was first synthesised in the early 1930s by Ernest H. Volwiler and Donalee L. Tabern at Abbott Laboratories in Illinois. Abbott manufactured the drug until 2004, when its Hospital Products division was spun off as Hospira. Following difficulties in obtaining raw materials, Hospira ceased manufacturing the drug in the U.S. in 2010, resulting in a world shortage, and bringing a darker side of thiopental sodium to public prominence. For in over 30 states of the U.S., thiopental sodium is used as part of the protocol for capital punishment by lethal injection. An initial injection of a very large dose of the drug is used to ensure rapid loss of consciousness, and is followed sequentially by injection of the muscle relaxant pancuronium bromide, which causes paralysis and stops breathing, and potassium chloride, which stops the heart. The states of Ohio and Washington have used thiopental sodium alone in cases where the normal procedure failed due to the inability to locate suitable veins. The shortage of thiopental sodium has therefore caused problems for the U.S. prison authorities, who have been looking abroad for supplies. For Arizona State Prison Complex, Florence, success was found in the U.K. last September: in Horn Lane, Acton, in west London, a small office is home to a driving school and a pharmaceutical wholesalers named Dream Pharma Ltd. [5], who supplied thiopental sodium apparently obtained from the British license holder, Archimedes Pharma UK [6]. Following a public outcry and a legal challenge, the British government reversed an initial decision and imposed export controls on the drug in late November 2010 to prevent its use in executions.
Hospira was originally planning to move production of thiopental sodium from the U.S. to Italy. However, the Italian parliament would only allow the drug to be made there if Hospira could guarantee that it would not be used in capital punishment. Since Hospira typically distributes the drug through wholesalers, the end use would be difficult to guarantee, so Hospira have recently decided to abandon attempts to resume production of the drug [6,7]. So now U.S. prison authorities must look elsewhere for supplies, or change to a different drug and face the inevitable legal challenges. In the meantime, it seems likely that the number of prisoners on "death row" in the U.S. (there are currently over 300 in Texas alone) will continue to rise.
Reference(s)
- Payne, J.P. (1994) Awareness and its medicolegal implications. Br. J. Anaesth., 73, 38–45.
- Bennetts, F.E. (1995) Thiopentone anaesthesia at Pearl Harbor. Br. J. Anaesth., 75, 366–368.
- Adams, R.C. and Gray, H.K. (1943) Intravenous anesthesia with Pentothal sodium in the care of gunshot wounds associated with accompanying severe traumatic shock and loss of blood: report of a case. Anesthesiology, 4, 70–73.
- Price, D. (2011) The case for sodium thiopental (letter) The Times, February 16 (Issue 70184), page 23.
- http://news.bbc.co.uk/today/hi/today/newsid_9342000/9342976.stm.
- http://www.guardian.co.uk/world/2011/jan/23/lethal-injection-sodium-thiopental-hospira?INTCMP=SRCH.
- Hospira Statement Regarding PentothalTM (sodium thiopental) Market Exit (Hospira news release), Jan. 21, 2011.

7 February 2011, Clenbuterol
One of the most recent high-profile allegations of the use of performance-enhancing drugs in sport involves the Spanish winner of the 2010 Tour de France cycle race, Alberto Contador. Following detection of a small amount of the sympathomimetic amine clenbuterol (CHEBI:174690) in a urine sample taken during a rest day of the 2010 Tour, Contador has been given a provisional one-year ban from racing by the Royal Spanish Cycling Federation and could be stripped of his 2010 Tour title by the Union Cycliste Internationale, the world governing body for cycle racing. Throughout the proceedings, Contador has continued to protest his innocence, claiming the source of the drug to have been contaminated meat.Clenbuterol is most commonly available as its hydrochloride salt and is used principally by sufferers of breathing disorders as a decongestant and bronchodilator. It is a β-adrenergic agonist with some structural and pharmacological similarities to adrenaline (epinephrine) and albuterol (salbutamol). Its effect in increasing the rate at which body fat is metabolised has led to its use in livestock to increase the muscle-to-body ratio and thus obtain leaner meat, a practice that is banned in the European Union and many other countries. In the USA, incidences of localised food contamination led its use being restricted in 2006 to the treatment of horses, while in China two cases of widespread poisoning have occurred, thought to be due to the consumption of meat infected with clenbuterol [1,2].
Reference(s)
2010
6 December 2010, Coenzyme Q10
Coenzyme Q10 (ChEBI:46245, also known as ubiquinone-10, CoQ10, CoQ, Q10 or simply Q) is a ubiquinone containing 10 isoprenoid units. First discovered in 1957 by Crane et al. [1], its chemical structure was determined by Karl Folkers [2], who later won the Priestley medal from the American Chemical Society. This oil-soluble, vitamin-like micronutrient forms part of the electron transport chain which, in the process of aerobic respiration, generates 95% of the human body's energy as ATP [3].Coenzyme Q10 is synthesized de novo by every cell in the body, but levels decrease with age, in several clinical disorders, and in patients administered certain drugs such as hydroxymethylglutaryl-CoA reductase inhibitors (commonly known as statins). With cardiovascular disease being a leading cause of death in the West, evidence that oral supplements of coenzyme Q10 can benefit patients suffering from heart disease is of increasing appeal. Evidence is also accumulating for its effective treatment of other ailments including mitochondrial disorders and neurodegenerative diseases such as amyotrophic lateral sclerosis (ALS), Huntington's disease and Parkinson's disease.
Coenzyme Q10 is one of the best-selling dietary supplements worldwide, available over the counter from health food shops and pharmacies. Its popularity may be due to the wide-ranging claims made for its effectiveness in a myriad of human health issues: it is marketed as an energy booster; a periodontal health promoter; an agent for maintaining normal blood-cholesterol levels; an enhancer of cognitive function; a remedy for hypertension, migraine headaches, radiation injury and cancer; and a superdrug capable of delaying or even reversing the effects of aging. However, perusal of the scientific literature reveals that, while data supporting some claims are forthcoming (such as in the case of heart disease and mitochondrial function), coenzyme Q10 is neither panacea nor elixir [4,5].
Reference(s)
- Crane, F.L., Hatefi, Y., Lester, R.L. and Widmer, C. (1957) Isolation of a quinone from beef heart mitochondria. Biochim. Biophys. Acta 25, 220–221.
- Wolf, D.E., Hoffman, C.H., Trenner, N.R., Arison, B.H., Shunk, C.H., Linn, B.O., McPherson, J.F. and Folkers, K. (1958) Coenzyme Q. I. Structure studies on the coenzyme Q group. J.Am. Chem. Soc. 80, 4752.
- Ernster, L. and Dallner, G. (1995) Biochemical, physiological and medical aspects of ubiquinone function. Biochim. Biophys.Acta 1271, 195–204.
- Watts, T.L. (1995), Coenzyme Q10 and periodontal treatment: is there any beneficial effect? Br. Dent. J. 178, 209–213.
- European Food Safety Authority Panel on Dietetic Products, Nutrition and Allergies (2010), Scientific Opinion on the substantiation of health claims related to coenzyme Q10 and contribution to normal energy-yielding metabolism (ID 1508, 1512, 1720, 1912, 4668), maintenance of normal blood pressure (ID 1509, 1721, 1911), protection of DNA, proteins and lipids from oxidative damage (ID 1510), contribution to normal cognitive function (ID 1511), maintenance of normal blood cholesterol concentrations (ID 1721) and increase in endurance capacity and/or endurance performance (ID 1913) pursuant to Article 13(1) of Regulation (EC) No 1924/2006. EFSA J. 8, 1793–1819.
1 November 2010, Graphene
The discovery of graphene (ChEBI:36973) has unusual origins in pencil lead and adhesive tape. In 2004, two scientists at the University of Manchester, UK attached adhesive tape to a piece of graphite then repeatedly peeled it back until they had the thinnest layer possible [1]. This was then attached to an oxidised silicon plate and the ultrathin layers observed using an electron microscope. Because graphene is so thin, it is virtually transparent and is the strongest material known to science [2]. It conducts electricity better than copper and also outperforms all other known materials as a conductor of heat.The two scientists, Andre Geim and Konstantin Novoselov have shared the 2010 Nobel Prize for Physics for their "groundbreaking experiments regarding the two-dimensional material graphene".
The breakthrough has led to an explosion of interest in graphene. The best-known potential use for graphene is as a replacement for silicon in computer chips. Graphene's electrons move 100 to 1,000 times faster than those of silicon meaning less power will be required for the same computing capacity. this could result in a new generation of faster smaller computers. The incredible strength of graphene provides the possibility of mixing small amounts into other materials and using the resulting composites to produce stronger and lighter materials. The major hold-up for progress is finding a way to produce graphene on an industrial scale. The process using adhesive tape and graphite known as 'exfoliation' is slow and labour intensive but has been scaled up with some companies currently able to produce graphene by the ton [3].
Reference(s)
- Novoselov, K.S., Geim, A.K., Morozov, S.V., Jiang, D., Zhang, Y., Dubonos S.V., Grigorieva I.V. and Firsov, A.A. (2004), Electric field effect in atomically thin carbon films. Science 306, 666–669.
- Lee, C., Wei X., Kysar, J.W. and Hone J. (2008) Measurement of the elastic properties and intrinsic strength of monolayer graphene. Science 321, 385–388.
- Segal, M. (2009) Selling graphene by the ton. Nature Nanotechnology 4, 612–614.
4 October 2010, Aldicarb
Aldicarb (CHEBI:2555) is a systemic insecticide, acaricide (a substance that kills ticks and mites), and nematocide (a substance that kills parasitic nematodes). It is commonly used under the trade name Temik, a granular mix containing 10–15% of the active ingredient, which is a mixture of E and Z isomers (it is not certain which is the more active).After the granules are applied to the soil, aldicarb dissolves in water present in the soil and is then taken up by the roots of the plants being protected. First described in the 1960s [1,2], it was introduced to the market in 1970 by Union Carbide Corporation and has since been used for the control of pests on a wide variety of crops, including bananas, beans, carrots and parsnips, citrus, cotton, onions, pecans, potatoes, sorghum, soya, sugar beet, and sweet potatoes.
One of the carbamate class of pesticides, aldicarb is an acetylcholinesterase inhibitor. It thus acts as a nerve poison by disrupting nerve impulses. Although not thought to be teratogenic, mutagenic or carcinogenic, it is the most toxic widely used pesticide. LD50 values for aldicarb administered in liquid form to rats, mice, and rabbits range from just 0.5 to 1 mg/kg; though less poisonous, the granules are still classed as highly toxic [3]. Moreover, because of its moderate solubility in water it can leach through soil into groundwater, and thus, perhaps unsurprisingly, its use has been beset by controversy.
Aldicarb is manufactured by the addition of 2-methyl-2-(methylsulfanyl)propanaldoxime to methyl isocyanate. In 1984, a leak of methyl isocyanate at a Union Carbide factory in Bhopal, India, making aldicarb and the related pesticide carbaryl for the Indian cotton crop, killed over 3,000 local residents; some estimates of the death toll exceed 15,000 [4]. (Union Carbide subsequently sold its worldwide agrochemical division to Rhône-Poulenc, who became part of Aventis CropScience GmbH in 1999 and were in turn acquired by Bayer in 2002 to form part of Bayer CropScience AG, the current manufacturer). In the U.S. in the mid-1980s, several incidents in which misapplication of aldicarb resulted in contaminated cucumbers and watermelons occurred, causing adverse effects in people. Subsequent reviews resulted in reductions in the levels of permitted residues and increased restrictions on the conditions under which use of aldicarb was allowed. Despite protests from farmers that alternative products were much less effective, its use was effectively banned in Europe in 2007 [4].
Now, 40 years after it was first registered, aldicarb is approaching the end of the road. Bayer CropScience has recently announced that it is to remove it voluntarily from the market worldwide by the end of 2014. In an agreement with the U.S. Environmental Protection Agency [5], U.S. farmers must stop using aldicarb on citrus and potatoes by the end of 2011, and on other crops by September 2018.
Reference(s)
- Payne, L.K. Jr and Weiden, M.H.J. (1965) 2-Hydrocarbylthio-sulfinyl and sulfonylalkanal carbamoyloximes. U.S. Pat., US3217037.
- Payne, L.K. Jr, Stansbury H.A. Jr and Weiden, M.H.J. (1966) The synthesis and insecticidal properties of some cholinergic trisubstituted acetaldehyde O-(methylcarbamoyl)oximes. J. Agric. Food Chem., 14, 356–365.
- Risher, J.F., Mink, F.L. and Stara, J.F. (1987) The toxicologic effects of the carbamate insecticide aldicarb in mammals: a review. Environ. Health Perspect., 72, 267–281.
- http://www.pesticides.gov.uk/approvals.asp?id=2065
- U.S. Environmental Protection Agency (2010) Agreement to Terminate All Uses of Aldicarb, http://www.epa.gov/oppsrrd1/REDs/factsheets/aldicarb_fs.html
6 September 2010, Melamine
The search for a rapid and effective test for melamine (CHEBI:27915) became important following the contaminated milk scandal in China in 2008, when milk powder deliberately tainted with melamine killed six infants and sickened an estimated further 300,000.Melamine (1,3,5-triazine-2,4,6-triamine) is an organic base, produced as a white, crystalline powder. It is only slightly soluble in water and is usually used to make melamine formaldehyde resin, fertilizer, flame retardants and other products. Its use as an additive in food or related ingredients is prohibited, as excessive ingestion of melamine will cause insoluble melamine cyanurate crystals to form in the kidneys leading to acute renal failure. Melamine's high nitrogen content (66% N by mass) gives it the analytical characteristics of protein molecules in some simple tests for protein used in the food industry, and it is this feature that has led to its illegal use as a food adulterant
Currently the most common tests for melamine are liquid chromatography–tandem mass spectrometry (LC–MS/MS) and gas chromatography–mass spectrometry (GC–MS). Other tests have been developed, e.g. capillary zone electrophoresis (CZE) and micellar electrokinetic chromatography (MEKC), but all of these tests, although highly sensitive, are time-consuming and labour-intensive. Now, Chinese scientists funded by the Chinese Ministry of Health have announced in the peer-reviewed journal Talanta that they have developed a quick and simple colorimetric testing method to detect melamine in milk products [1]. The method uses gold nanoparticles which, as they approach each other and aggregate, experience a colour change from wine-red to purple or blue. The presence of melamine in the tested product causes this aggregation approach to commence. The new test, which can be completed within 30 minutes, is low-cost, is visible to the naked eye and requires no pre-treatment, can be used on-site to test for melamine in both infant formula and liquid milk, with sufficient sensitivity for detection at the levels required by regulatory bodies.
Reference(s)
- Guo, L., Zhong, J., Wu, J., Fu, F, Chen, G. and Lin, S. (2010) Visual detection of melamine in milk products by label-free gold nanoparticles. Talanta, doi:10.1016/j.talanta.2010.07.035.
2 August 2010, Oxytocin
Oxytocin (ChEBI:7872), sometimes known as the "love molecule" or the "trust molecule" plays an important role in many processes. These include uterine contractions during childbirth, sexual arousal, lactation, puberty, orgasm, facial recognition, trust, memory formation and pair bonding.Oxytocin is a cyclic peptide hormone with just nine amino acids in sequence (CYIQNCPLG) that also acts as a neurotransmitter in the brain [1] where it is produced in the hypothalamus. It was the first ever polypeptide hormone to be sequenced and synthesized biochemically [2], work for which the American biochemist Vincent du Vigneaud was awarded the 1955 Nobel Prize in Chemistry.
Together with the neuropeptide argipressin (arginine vasopressin), it is believed to influence social cognition and behaviour. First shown in mice, recent studies have shown that also in humans simply sniffing a spray containing oxytocin increases a person's level of trust in others. [3].
Reference(s)
- Lee, H.J., Macbeth, A.H., Pagani, J.H. and Young, W.S. (2009) Oxytocin: the great facilitator of life. Prog. Neurobiol. (Amsterdam, Neth.) 88, 127–151.
- du Vigneaud, V., Ressler, C., Swan, J.M., Roberts, C.W., Katsoyannis, P.G. and Gordon, S. (1953) The synthesis of an octapeptide amide with the hormonal activity of oxytocin. J. Am. Chem. Soc. 75, 4879–4880..
- Kosfeld, M., Heinrichs, M., Zak, P.J., Fischbacher, U. and Fehr, E. (2005) Oxytocin increases trust in humans. Nature 435, 673–676.
5 July 2010, Edaxadiene
Tuberculosis is a contagious pulmonary disease that is on the increase, killing 1.5 to 2 million people worldwide annually. It is caused by the pathogen Mycobacterium tuberculosis, but details about the virulence and infectivity of this organism are still only partly understood. This has in turn slowed progress towards the development of new drugs to combat the disease.In 2009, a team of scientists from Iowa State, Illinois and Cornell Universities led by Professor Reuben Peters isolated from M. tuberculosis a halimane-type diterpenoid which they named edaxadiene, [1]. They found that the compound was produced by M. tuberculosis as a defence mechanism against attack by human macrophage cells, cells which aim to engulf and destroy infectious microbes. Peters' team proposed for the new molecule a structure consisting of a tricyclic core with five adjacent stereocentres.
Elsewhere, during an attempt to synthesise edaxadiene so as to be able to study it in more depth, Erik Sorensen and his colleagues at Princeton University deduced on the basis of spectral evidence that the molecule contained only a bicyclic core and had a longer side-chain. This alternative structure for edaxadiene, now confirmed following a successful synthesis of the molecule by Sorensen's team [2], forms our Entity of the Month and is shown as CHEBI:59685 In fact, a molecule with this same structure had been isolated some 6 years earlier from a sponge, Raspailia sp., collected from the Nosy Be island of Madagascar by a team led by Yoel Kashan of Tel-Aviv University and named appropriately by them as nosyberkol, [3].
The correct identification and successful synthesis of edaxadiene/nosyberkol should enable sufficient quantities of this compound to be prepared in order to elucidate more precisely its role in the infectivity and virulence of M. tuberculosis. It thus looks set to make a significant impact on our understanding of, and ability to treat, tuberculosis.
The background image shows a close-up of a culture of M. tuberculosis, revealing the organism's colonial morphology.
Reference(s)
- Mann, F.M., Prisic, S., Hu, H., Xu, M., Coates, R.M. and Peters, R.J. (2009), Characterization and inhibition of a Class II diterpene cyclase from Mycobacterium tuberclosis - Implications for tuberculosis. J. Biol. Chem., 284, 23574–23579.
- Spangler, J.E., Carson, C.A. and Sorensen, E.J. (2010) Synthesis enables a structural revision of the Mycobacterium tuberculosis-produced diterpene, edaxadiene. Chem. Sci., DOI: 10.1039/c0sc00284d.
- Rudi, A., Aknin, M., Gaydou, E. and Kashman, Y. (2004) Asmarines I, J, and K and nosyberkol: four new compounds from the marine sponge Raspailia sp. J. Nat. Prod., 67, 1932–1935.
2 June 2010, 6-acetyl-2,3,4,5-tetrahydropyridine
Chemistry, like most other fields of human endeavour, has a tremendous capacity for both good and evil. However, arguably one of the best and most delightful reactions in chemistry is the Maillard reaction. It occurs when amino acids are heated together with sugar and is therefore a prominent reaction when baking bread or brewing beer: many of the reaction products provide the characteristic flavours of these foods, which we all enjoy so much.While the chemical structures and identities of most of the products of this form of "non-enzymatic browning" are only poorly characterised or unknown, our Entity of the Month 6-acetyl-2,3,4,5-tetrahydropyridine (CHEBI:59533) is an exception. It is a well known aromatic compound, which is responsible for the flavour of white bread, popcorn and tortillas and has an extremely low odour threshold, between 0.02 and 0.06 ng l–1.[1] It exists in a tautomeric equilibrium with 6-acetyl-1,2,3,4-tetrahydropyridine, the two forms usually occurring in foods in a 1:2 ratio.The compound can be synthesized in a simple three-step procedure. In a first step, BOC-protected 2-piperidone is treated with 1-ethoxy-1-lithioethene in a bid to build up the acetyl side-chain. This results in ring opening and the formation of a linear ketone which, after treatment with toluene-p-sulfonic acid, reforms the heterocycle in the form of an ene-carbamate. Treatment of the latter with potassium hydroxide yields the final product [1].
The Maillard Reaction is named after Louis Camille Maillard, a precocious French physiologist, who first described it in the 1910s. Maillard is also known for his contributions towards the diagnosis of kidney disorders.
Our background image shows freshly toasted bread – and the brown colour (the Maillard reaction is a method for non-enzymatic browning) is indicative of the reaction having taken place.
Reference(s)
- Harrison, T.J. and Dake, G.R. (2005) An expeditious, high-yielding construction of the food aroma compounds 6-acetyl-1,2,3,4-tetrahydropyridine and 2-acetyl-1-pyrroline. J. Org. Chem. 70, 10872–10874.
5 May 2010, Mephedrone
Mephedrone (CHEBI:59331) is a synthetic central nervous system stimulant and entactogen drug chemically related to cathinone, the psychoactive alkaloid present in the khat plant (Catha edulis, family Celastraceae). It can be synthesised from 4-methylpropiophenone by an initial bromination at the β-carbon followed by replacement of the bromine by a methylamino group derived from methylamine hydrochloride. Although it was probably not available until 2007, by 2009 mephedrone had become the fourth most popular street drug in the UK, behind cannabis, cocaine and ecstasy. Little is currently known regarding its pharmacology or toxicology, although one recent report suggests the likelihood that it stimulates the release of, and then inhibits the reuptake of, monoamine neurotransmitters [1].Although already listed as a prohibited substance in many countries, in others it has varying degrees of legality (notably the USA where it is currently unscheduled under the Controlled Substances Act). In the UK, a decision by the Home Secretary to classify mephedrone as illegal caused the resignation of two members of the Advisory Council on the Misuse of Drugs (ACMD) which has led in turn to a general questioning of UK drugs policy. Mephedrone finally became classified as a Class B drug in the UK on April 16, 2010 – prior to this time it was often sold openly under the guise of a 'plant food' (although having no known use as such).
Reference(s)
- Winstock, A., Marsden, J. and Micheson, L. (2010). What should be done about mephedrone? BMJ [Br. Med. J.] 340, c1605.
7 April 2010, 8-OHdG
8-Hydroxy-2'-deoxyguanosine (8-OHdG, ChEBI:40304) is an important molecule in oxidative stress used as a biomarker of many processes involving reactive oxygen species. Also known as 8-oxo-dG (this abbreviation derived from its tautomeric name 8-oxo-7,8-dihydro-2'-deoxyguanosine) and as HMDB03333 in the Human Metabolome Database [1], it has been used especially as a sensitive marker of the DNA damage caused by hydroxyl radical attack at C-8 of guanine. This damage, if left unrepaired, has been proposed to contribute to mutagenicity and cancer promotion [2]. This use of 8-OHdG as a biomarker for DNA damage extends over a wide range of scenarios [3,4,5,6], because it is one of the major products of DNA oxidation.More recent work by Junko Fujihara and his colleagues at Shimane University in Japan has demonstrated how 8-OHdG can be used as a possible marker for arsenic poisoning, since antiquity a method of dispatch frequent in homicide and suicide cases [7]. Fujihara's study however focuses principally on the use of arsenic in medicine, and specifically in demonstrating a relationship between concentrations of 8-OHdG and various arsenic compounds in the urine of a patient with acute promyelocytic leukaemia being treated with arsenic trioxide. Their conclusions that 8-OHdG in urine can be used therapeutically as a key biomarker for arsenic compounds may also find application in the diagnosis of arsenic poisoning when arising from the consumption of seafood such as fish, shrimp, oysters and seaweeds, organisms known to contain appreciable amounts of arsenic compounds.
Reference(s)
- Wishart, D.S., Knox, C., Guo, A.C., Eisner, R., Young, N., Gautam, B., Hau, D.D., Psychogios, N., Dong, E., Bouatra, S. et al. (2009). HMDB: a knowledgebase for the human metabolome. Nucleic Acids Res. 37 (Database issue), D603–610.
- Kuchino, Y., Mori, F., Kasai, H., Inoue, H., Iwai, S., Miura, K., Ohtsuka, E. and Nishimura, S. (1987). Misreading of DNA templates containing 8-hydroxydeoxyguanosine at the modified base and at adjacent residues. Nature 327 (6117), 77–79.
- Wu, L.L., Chiou, C.C., Chang, P.Y. and Wu, J.T. (2004). Urinary 8-OHdG: a marker of oxidative stress to DNA and a risk factor for cancer, atherosclerosis and diabetics. Clin. Chim. Acta339, 1–9.
- Schriner, S.E., Linford, N.J., Martin, G,M., Treuting, P., Ogburn, C.E., Emond, M., Coskun, P.E., Ladiges, W., Wolf, N., Van Remmen, H. et al. (2005). Extension of murine life span by overexpression of catalase targeted to mitochondria. Science 308, 1909–1911.
- Sumida, S. et al (1997). Effect of a single bout of exercise and beta-carotene supplementation on the urinary excretion of 8-hydroxy-deoxyguanosine in humans. Free radical research 27 (6), 607-618. PMID:9455696
- Tarng, D.-C., Huang, T.-P., Wei, Y.-H., Liu, T.-Y., Chen, H.-W., Chen, T.W. and Yang, W.-C. (2000). 8-Hydroxy-2'-deoxyguanosine of leukocyte DNA as a marker of oxidative stress in chronic hemodialysis patients. Am. J. Kidney Dis. 36, 934–944.
- Fujihara, J., Agusa, T., Tanaka, J., Fujii, Y., Moritani, T., Hasegawa, M., Iwata, H., Tanabe, S. and Takeshita, H. (2009).8-Hydroxy-2'-deoxyguanosine (8-OHdG) as a possible marker of arsenic poisoning: a clinical case study on the relationship between concentrations of 8-OHdG and each arsenic compound in urine of an acute promyelocytic leukemia patient being treated with arsenic trioxide. Forensic Toxicol. 27, 41–44.
3 March 2010, Sildenafil citrate
Few chemical compounds are better known to the general public than sildenafil citrate (CHEBI:58987), traded under the name of "Viagra".The compound was first synthesised by chemists working at Pfizer, with a view to using it for the treatment of hypertension and angina pectoris. Whilst having been found to be ineffective against angina in clinical trials, it has been observed to induce penile erections and was therefore marketed by Pfizer as a drug for the treatment of erectile dysfunction. A number of synthetic routes for the preparation of the parent sildenafil have been reported [1]. A common industrial synthetic route is through reaction of 4-amino-1-methyl-3-N-propylpyrazole-5-carboxamide and 2-ethoxy-5-(4-methylpiperazin-1-yl)sulfonylbenzoic acid followed by subsequent cyclisation to sildenafil through heating under acidic conditions.Sildenafil has been shown to be an inhibitor of cyclic guanosine monophosphate specific phosphodiesterase type 5, an enzyme which is responsible for the degradation of 3',5'-cyclic GMP (cyclic guanosine monophosphate, cGMP) in the corpus cavernosum. This leads to the presence of increased levels of cGMP, which, in turn causes vasodilation of the helicine arteries and thus increased blood flow into the spongy tissue of the penis [2].
Apart from the treatment of sexual dysfunction, sildenafil is also used in the treatment of pulmonary arterial hypertension and works again through relaxation of the arterial wall, which leads to a decrease in arterial resistance [3]. Furthermore – and arguably most interestingly – sildenafil has been found to decrease the time necessary for the re-entrainment of circadian rhythms after phase advances in the light–dark cycle (such as occur on transmeridian eastbound flights) in members of the Cricetidae family [4]. The discovery was rewarded with the award of an Ig Nobel Prize in Aviation in 2007.
Creative Commons licensed picture of Sildenafil crystals taken from Wellcome Trust Images.
Reference(s)
- Dunn, P.J. (2005) Synthesis of commercial phosphodiesterase(v) inhibitors. Org. Process Res. Dev. 9, 88–97.
- David, J.W., Stephen, F., Michael, J.A. and Gary, J.M. (1999) Sildenafil citrate and blood-pressure-lowering drugs: results of drug interaction studies with an organic nitrate and a calcium antagonist. Am. J. Cardiol. 83 (5, Suppl. 1), 21–28.
- Richalet, J.-P., Gratadour, P., Robach, P., Pham, I., Dechaux, I., Joncquiert-Latarjet, A., Mollard, P., Brugniaux, J. and Cornolo, J. (2005) Sildenafil inhibits altitude-induced hypoxemia and pulmonary hypertension. Am. J. Respir. Crit. Care Med. 171, 275–281.
- Agostino, P.V., Plano, S.A. and Golombek, D.A. (2007) Sildenafil accelerates reentrainment of circadian rhythms after advancing light schedules. Proc. Natl. Acad. Sci. U. S. A. 104, 9834–9839.
3 February 2010, Ferrocene
Ferrocene (CHEBI:30672) is not only an intensely useful chemical entity which has found applications in such diverse fields such as anti-cancer therapy and materials science, but it has also brought together a number of the most eminent chemical researchers of the century and led to the discovery of a new type of bonding. The orange compound was, as is often the case, discovered accidentally by Pauson (of Pauson–Khand reaction fame) and Kealy, who attempted synthesis of fulvalene through oxidative coupling of cyclopentadienyl magnesium bromide and iron(III) chloride [1]. The result was an orange powder, which proved to be extremely stable and whose structure remained, for a while at least, a mystery: Pauson and Khand initially suggested that two cyclopentadienyl moieties, which were bound to the iron(II) in an η1 fashion rather than in the η5-motif we know to be correct today. The new bonding motif caused a little sensation in the structural chemistry community when it was postulated [2].The correct metallocenic structure was independently determined/deduced by Woodward and Wilkinson [3] as well as by E.O. Fischer [4], who subsequently extended the series of known metallocene compounds considerably. Significant amounts of work were also done by Myron Rosenblum, Woodward's student and one of the co-authors on their original paper reporting the ferrocene structure. Many accounts of the beginning and further development of ferrocene research have since been written [5–9].
Ferrocene is a remarkably stable organometallic compound and shows chemistry which is very similar to that of 'classical' aromatic compounds. It readily undergoes Friedel-Crafts chemistry [10] and can also be lithiated [11], thus showing a wide spectrum of interaction with both electrophiles and nucleophiles. This, in turn, means that a large number of derivatives can be prepared, which opens the material up to a wide range of applications. Chemical examples include the use of ferrocene as a backbone in ligand systems and as an oxidising agent. Ferrocene can be polymerised through derivatisation to vinylferrocene and subsequent polymerisation to give poly(vinylferrocene) [12].
The compound has use as an anti-knock agent, replacing the much more toxic tetraethyllead. Furthermore, ferrocenyl analogues of tamoxifen, an estrogen receptor antagonist, have also been reported in the literature [13].
Reference(s)
- Kealy, T.J. and Pauson, P.L. (1951) A new type of organo-iron compound. Nature 168, 1039–1040.
- Lazlo, P. and Hoffmann, R. (2000) Ferrocene: Iron-clad history or Rashomon Tale? Angew. Chem., Int. Ed. 39, 123–124.
- Wilkinson, G., Rosenblum, M., Whiting, M.C. and Woodward, R.B. (1952) The structure of iron bis-cyclopentadienyl. J. Am. Chem. Soc. 74, 2125–2126.
- Fischer, E.O. and Pfab, W. (1952) Zur Kristallstruktur der Di-Cyclopentadienyl-Verbindungen des zweiwertigen Eisens, Kobalts und Nickels. Z. Naturforsch., B 7, 377–379.
- Fischer, E.O. and Jira, R. (2001) How metallocene chemistry and research began in Munich. J. Organomet. Chem. 637–639, 7–12.
- Pauson, P.L. (2001) Ferrocene—how it all began. J. Organomet. Chem. 637–639, 3–6.
- Rosenblum, M. (2001) The early ferrocene days—a personal recollection. J. Organomet. Chem. 637–639, 13–15.
- Whiting, M.C. (2001) Recollections of the arrival of ferrocene. J. Organomet. Chem.637–639, 16–17.
- Cotton, F.A. (2001) Cyclopentadienyl–metal chemistry in the Wilkinson Group, Harvard, 1952–1955. J. Organomet. Chem. 637–639, 18–26.
- Wang, R., Hong, X. and Shan, Z. (2008) A novel, convenient access to acyl ferrocenes: acylation of ferrocene with acyl chlorides in the presence of zinc oxide. Tetrahedron Lett. 49, 636–639 and references therein.
- Ueberbacher, B.J., Griengl, H. and Weber, H. (2008) Ortho-lithiation of free ferrocenyl alcohols: a new method for the synthesis of planar chiral ferrocene derivatives. Chem. Commun., 3287–3289.
- Hudson, R.D.A. (2001) Ferrocene polymers: current architectures, syntheses and utility. J. Organomet. Chem. 637–639, 47–69.
- Top, S., Vessieres, A., Leclercq, G., Quivy, J., Tang, J., Vaissermann, J., Huche, M. and Jaouen, G. (2003) Synthesis, biochemical properties and molecular modelling studies of organometallic specific estrogen receptor modulators (SERMs), the ferrocifens and hydroxyferrocifens: evidence for an antiproliferative effect of hydroxyferrocifens on both hormone-dependent and hormone-independent breast cancer cell lines. Chem.—Eur. J. 9, 5223–5236.
6 January 2010, (+)-Abscisic acid
(+)-Abscisic acid (CHEBI:2365), known commonly just as abscisic acid or ABA, is a ubiquitous isoprenoid plant hormone which is synthesized in the methylerythritol phosphate (MEP) pathway (also known as the non-mevalonate pathway) by cleavage of C40 carotenoids. First identified and characterised in 1963 by Fredrick Addicott and his associates at the University of California, Davis [1], ABA was originally believed to play a major role in abscission of fruits (hence its early name of 'abscisin II'). This is now known to be true for only a small number of plants, a wider role being to act as a regulator of plant responses to a variety of environmental stresses such as drought, extremes of temperatures, and high salinity. Such responses include stimulating the closure of stomata, inhibiting shoot growth while not affecting root growth, and inducing seeds to synthesise storage proteins.Because of its essential function in plant physiology, targeting the ABA signalling pathway holds considerable promise for future applications in agriculture. Now, in a recent issue of Nature, Ning Zheng and his co-worker Laura Sheard from the University of Washington summarise recent converging studies which reveal the details of how ABA transmits its message [2]. In particular, an article by an international team led by Eric Xu of the Van Andel Research Institute describes how their crystallographic work on unbound ABA and ABA bound to some of its receptors, together with extensive biochemical studies from elsewhere, identify a conserved gate–latch–lock mechanism underlying ABA signalling [3].
Reference(s)
- Okhuma, K., Lyon, J.L., Addicott, F.T. and Smith O.E. (1963) Abscisin II, an abscission-accelerating substance from young cotton fruit. Science 142, 1592–1593.
- Sheard, L.B. and Ning Zheng (2009) Plant biology: Signal advance for abscisic acid. Nature 462, 575–563.
- Melcher K. et al. (2009) A gate–latch–lock mechanism for hormone signalling by abscisic acid receptors. Nature 462, 602–608.
2009
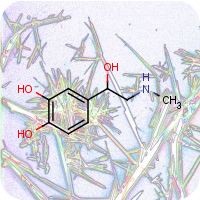
3 December 2009, Adrenaline
Adrenaline (CHEBI:33568), also known as epinephrine, is a catecholamine that acts as a hormone and neurotransmitter. It was first isolated from an extract of the suprarenal (adrenal) gland as its mono-benzoyl derivative by the American biochemist and pharmacologist John Jacob Abel in 1889 [1] who later also crystallised it as a hydrate. The pure compound was produced in 1901 by the Japanese industrial chemist Jokichi Takamine [2] and patented as 'Adrenalin'. Two chemists, Stolz and Dakin, independently reported the synthesis of the compound in 1904 [3,4].Adrenaline is a potent 'fight-or-flight' hormone, which is produced in stress situations. When produced in the body, it leads to an increase in heart-rate, vasodilation and the supply of both glucose and oxygen to the muscles and the brain, thus preparing the body for rapid action if needed. The increase in glucose supply is achieved through the binding of adrenaline to β-adrenergic receptors in the liver. This triggers the adenylate cyclase pathway, which, in turn, leads to increased glycogenolysis activity. On the other hand, adrenaline suppresses both digestive processes as well as immune responses. As such, it can be used in the treatment of anaphylactic shock [5] as well as for the treatment of cardiac arrest and cardiac disrythmias [6].
The biosynthesis of adrenaline is regulated by the central nervous system. It is ultimately derived from L-tyrosine, which is converted into L-dihydroxyphenylalanine (L-DOPA) by the action of tyrosine 3-monooxygenase (EC 1.14.16.2). Adrenaline is produced through the conversion of L-DOPA into dopamine into noradrenaline into adrenaline itself. Adrenaline crystallises in star-shaped needles (see background picture).
Reference(s)
- Abel, J.J. (1899) Ueber den blutdruckerregenden Bestandtheil der Nebenniere, das Epinephrin. Z. Physiol. Chem. 18, 318–324.
- Takamine, J., (1902) The isolation of the active principle of the suprarenal gland. J. Physiol. 27 (Suppl), xxix–xxx.
- Stolz, F. (1904) Ueber Adrenalin und Alkylaminoacetobrenzkatechin. Ber. Dtsch. Chem. Ges. 37, 4149–4154.
- Dakin, H.D. (1905) The synthesis of a substance allied to noradrenaline. Proc. Roy. Soc. Lon. Ser. B 76, 491–497.
- Anchor, J. and Settipane, R.A. (2004) Appropriate use of epinephrine in anaphylaxis. Am. J. Emerg. Med. 22, 488–490
- Rainer, T.H. and Robertson, C.E. (1996) Adrenaline, cardiac arrest and evidence based medicine. J. Accid. Emerg. Med. 13, 234–237
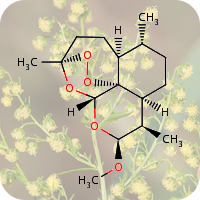
4 November 2009, Artemether
Artemether (CHEBI:195280) is a lipid-soluble antimalarial for the treatment of multi-drug resistant strains of Plasmodium falciparum malaria. First prepared in 1979 [1], it is a methyl ether of the naturally occurring sesquiterpene lactone (+)-artemisinin, which is isolated from the leaves of Artemisia annua L. (sweet wormwood), the traditional Chinese medicinal herb known as Qinghao. However, because of artemether's extremely rapid mode of action (it has an elimination half-life of only 2 hours, being metabolized to dihydroartemisinin which then undergoes rapid clearance), it is used in combination with other, longer-acting, drugs. One such combination, licensed in April of this year by the WHO, is Coartem in which the artemether is mixed with lumefantrine – a racemic mixture of a synthetic fluorene derivative known formerly as benflumetol – which has a much longer and pharmacologically complementary terminal half-life of 3–6 days, allowing the two drugs to act synergistically against Plasmodium.The molecule of artemether is interesting because of its extreme rigidity, with very few rotational bonds. Unlike quinine class antimalarial drugs, it has no nitrogen atom in its skeleton. However, an important chemical feature (and unique in drugs) is the presence of an O–O endoperoxide bridge which is essential for its antimalarial activity, as it is this bridge which is split in an interaction with heme, blocking the conversion into hemozoin and thus releasing into the parasite heme and a host of free radicals which attack the cell membrane.
Artemether is fully Rule-of-Five compliant and has recently also been under investigation as a possible candidate for cancer treatment [2,3].
Reference(s)
- Li, Y. et al. (1979) K'o Hsueh T'ung Pao, 24, 667 [Chem. Abstr., 91, 211376u (1979)].
- Singh, N.P. and Panwar, V.K. (2006) Case report of a pituitary macroadenoma treated with artemether. Integrative Cancer Therapies 5,391–394.
- Zhi-ping Wu et al. (2009) Inhibitive effect of artemether on tumor growth and angiogenesis in the rat C6 orthotopic brain gliomas model. Integrative Cancer Therapies 8, 88–92.
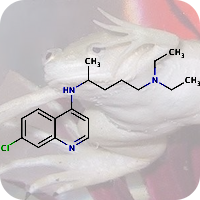
7 October 2009, Chloroquine
Chloroquine (CHEBI:3638) is a 4-aminoquinoline drug which has been used in the prevention or treatment of malaria since the 1930s. However, despite being originally cheap, safe and effective, its efficacy has declined gradually owing to the emergence and spread of chloroquine-resistant Plasmodium falciparum malaria parasites, such that in some areas of the world the drug is totally ineffective.P. falciparum feeds on haemoglobin protein, during the breakdown of which one or more of the various forms of the toxic and soluble molecule heme is produced. The parasite protects itself against destruction by the heme by biocrystallizing it to form the non-toxic pigment hemozoin which then collects in the organism's digestive vacuole. Chloroquine works by stopping this protective conversion of heme, causing a buildup of toxic iron waste which eventually causes cell lysis and death of the parasite.
In 2000 malarial resistance to chloroquine was linked to mutations in the P. falciparum Chloroquine Resistance Transporter (PfCRT) gene [1,2], but just how the PfCRT protein caused the resistance remained unclear. Now Dr Rowena Martin and her colleagues at the Australian National University, reporting in the journal Science [3], have studied PfCRT's function by isolating it in unfertilised eggs of the African Clawed Frog (Xenopus laevis). Martin's team were then able to show that PfCRT works by transporting the chloroquine out of the parasite's digestive vacuole, preventing it from reaching the concentrations it needs to work. Furthermore, tests with verapamil, a known 'resistance reversing' drug known to stop PfCRT's effects, showed that this actually blocked the chloroquine exit route, raising the possibility of producing future combinations of drugs of this type for more effective malarial treatments.
Reference(s)
- Fidock, D.A. et al. (2000) Mutations in the P. falciparum digestive vacuole transmembrane protein PfCRT and evidence for their role in chloroquine resistance. Mol. Cell 6, 861–871.
- Sidhu, A.B., Verdier-Pinard, D. and Fidock, D.A. (2002) Chloroquine resistance in Plasmodium falciparum malaria parasites conferred by pfcrt mutations. Science 298 , 210–213.
- Martin, R.E., Marchetti, R.V., Cowen, A.I., Howitt, S.M., Bröer, S. and Kirk, K. (2009) Chloroquine transport via the malaria parasite's Chloroquine Resistance Transporter. Science 325, 1680–1682.
26 August 2009, Cocaine
The notion of us all carrying cocaine (ChEBI:27958) in our wallets, purses or packets may not be as outrageous or unlikely as we might think! In what researchers describe as the largest, most comprehensive analysis to date of cocaine contamination in banknotes, scientists are reporting that 95% of dollar bills in Washington DC bear traces of the illegal drug cocaine, with high levels also oberved in other big cities, such as Baltimore, Boston and Detroit [1]. Researchers at the University of Massachusetts in Dartmouth tested notes from more than 30 cities from five countries (United States, Canada, Brazil, China and Japan). "To my surprise, we're finding more and more cocaine in banknotes," said lead researcher Yuegang Zuo. He suggested the rise may be due to the economic downturn, "with stressed people turning to cocaine".The U.S. and Canada were found to have the highest levels, with an average contamination rate of between 85 and 90 percent, while China and Japan had the lowest, between 12 and 20 percent contamination [1]. In the U.S. the cleanest bills were collected from Salt Lake City, home of the Church of Jesus Christ of Latter-Day Saints, better known as the Mormons [1].
Scientists have known for years that paper money can become contaminated with cocaine during drug deals and directly via snorting cocaine through rolled bills. Large-scale contamination is thought to take place when the notes are stacked together in currency-counting machines. In the new study, Zuo and colleagues used a modified form of a gas chromatograph-mass spectrometer for their measurements [2]. This allowed a faster, simpler and more accurate measurement of cocaine contamination than other methods, without destroying the currency. Amounts found in the 234 U.S. banknotes analysed ranged from .006 micrograms (thousands of times smaller than a single grain of sand) to over 1,240 micrograms of cocaine, or the equivalent of about 50 grains of sand, per bill. Zuo downplayed any health or legal concerns linked to the handling of contaminated notes. "For the most part, you can't get high by sniffing a regular banknote, unless it was used directly in drug uptake or during a drug exchange", Zuo said. "It also won't affect your health and is unlikely to interfere with blood and urine tests used for drug detection". The study, Zuo said, could help increase public awareness of cocaine use and help curb its abuse by assisting law enforcement agencies and forensic specialists to identify how the drug is used in a given community.
References
- U.S. banknotes show cocaine traces. news.bbc.co.uk. 17 August 2009.
- Zuo Y,, Kai Zhang K., Wu J., Rego C. and Fritz J. (2008)
An accurate and non-destructive GC method for determination of cocaine on U.S. paper currency. J. Separation Sci. 31, 2444–2450.

29 July 2009, Copernicium
Thirteen years after its discovery [1], element 112 has finally been given the proposed name 'copernicium' (ChEBI:33517) with the symbol Cp, in honour of the astronomer Nicolaus Copernicus. Copernicus deduced that the planets revolved around the sun and finally refuted the then accepted belief that the Earth was the centre of the universe. His finding was pivotal in the discovery of gravitational force, and led to the conclusion that the stars are incredibly far away and the universe inconceivably large, as the size and position of the stars does not change even though the Earth is moving.The International Union of Pure and Applied Chemistry (IUPAC) will officially endorse the new element's name in January 2010 in order to give the scientific community time to discuss the suggested name 'copernicium' prior to the IUPAC naming. Currently the element has the IUPAC systematic element name of Ununbium and the temporary symbol Uub used until the element gets an accepted name. Scientists from the Centre for Heavy Ion Research (GSI) in Germany, led by Professor Sigurd Hofmann, discovered copernicium during fusion experiments in 1996. "After IUPAC officially recognised our discovery, we agreed on proposing the name because we would like to honour an outstanding scientist" said Professor Hofmann.
Element 112 is the heaviest element in the periodic table, 277 times heavier than hydrogen. It is produced by a nuclear fusion, when bombarding zinc ions on to a lead target [1] in a heavy ion accelerator. As the element decays after a split second [1], its existence can only be proved with the assistance of extremely fast and sensitive analysis methods. Isolation of an observable quantity has never been achieved and may well never be. Since 1981, scientists at GSI have discovered six new chemical elements carrying atomic numbers 107-112 and have named five of them: element 107 is called bohrium (Bh), 108 is hassium (Hs), 109 is meitnerium (Mt), 110 is darmstadtium (Ds) and element 111 is called roentgenium (Rg).
Reference
- Hofmann S., Ninov, V., Hessberger, F.P., Armbruster, P., Folger, H., Münzenberg, G., Schött, H.J., Popeko, A.G., Yeremin, A.V., Saro, S., Janik, R., and Leino M. (2006) The new element 112. Zeitsch rift für Physik:A Hadrons and Nuclei. 354, 229–230.
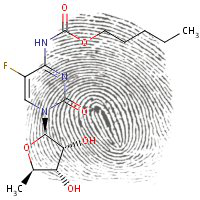
24 June 2009, Capecitabine
Capecitabine (CHEBI:31348) is a chemotherapy drug that is administered as a treatment for a variety of cancer types, including bowel cancer, stomach cancer, breast cancer and oesophageal cancer. It acts as a prodrug, undergoing a three-step enzymic conversion into 5-fluorouracil in the tumour, where it inhibits DNA synthesis and thus slows growth of tumour tissue. Capecitabine can be synthesized from the readily available starting materials D-ribofuranose and cytosine [1].When capecitabine is used for long-term treatment of recurrent cancers, one of the side-effects can be the onset of hand-foot syndrome, which eventually can lead to total eradication of the patients fingerprints [2]. This has recently proved rather inconvenient for one unsuspecting patient. A recent report [3] describes an instance where a patient on capecitabine for over three years went to the USA to visit relatives in December 2008. He was detained for 4 hours as his fingerprints could not be detected by immigration officials and was only allowed to enter after the officers were entirely satisfied that he posed no threat to security. As a result, all patients taking capecitabine long-term are being advised to travel with a letter from an oncologist stating condition and treatment being received to account for their lack of fingerprints.
As a final aside, recent reports suggest that the actual purpose of fingerprints is to enhance sensitivity rather than friction.
References
- Moon, B.S., Shim, A.Y., Lee, K.C., Lee, H.J., Lee, B.S., An, G.I., Yang, S. D., Chi, D.Y., Choi, C.W., Lim, S.M. and Chun, K.S. (2005) Synthesis of F-18 labeled capecitabine using [18F]F2 gas as a tumor imaging agent. Bull. Korean Chem. Soc. 26, 1865–1868.
- Chua, D., Wei, W.I., Sham, J.S.T. and Au, G.K.H. (2008) Capecitabine monotherapy for recurrent and metastatic nasopharyngeal cancer. Jpn. J. Clin. Oncol. 38, 244–249.
- Wong, M., Choo, S.-P. and Tan, E.-H. (2009) Travel warning with capecitabine. Annals of Oncology Advance Access May 26th 2009/doi:10.1093/annonc/mdp278.
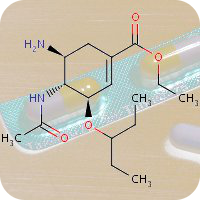
27 May 2009, Oseltamivir
Oseltamivir (CHEBI:7798) is an ethyl ester prodrug which requires hydrolysis to the corresponding carboxylate to be physiologically active. It is marketed as an antiviral drug for the treatment of both Influenzavirus A and Influenzavirus B infection in the form of its phosphate salt by Hofmann-La Roche (trade name Tamiflu). Oseltamivir inhibits the neuraminidase protein on the viral surface and interferes with virus particle aggregation and release.Oseltamivir is synthesized commercially from shikimic acid, the low availability of which is a major bottleneck in its production. Shikimic acid is only effectively isolated from Chinese star anise (Illicium verum) which is grown solely in four provinces of China and one in Vietnam. Some 90% of the annual harvest is used in the production of oseltamivir. For this reason, several alternative routes for shikimic acid production are being actively sought by a number of commercial companies. In addition, some alternative synthetic routes to oseltamivir which do not involve the intermediacy of shikimic acid have appeared in the literature [1–5].
The onset of the H5N1 avian influenza epidemic in Southeast Asia in 2005 led to fears of a pandemic and subsequent stockpiling of oseltamivir by several nations. However, although affluent countries like Japan, Britain and the USA are thought to have enough oseltamivir to reach about one quarter to a half of their populations, a recent press report highlights the situation with developing countries which may well have enough for only about 1 percent of their people or less. The 2009 outbreak in Mexico of a new strain of the influenza type A H1N1 virus (commonly called 'swine flu') has renewed fears of a pandemic and these, together with the high costs of stockpiling and maintaining expensive medicines for future epidemics that may never develop, have highlighted again the possibility that those countries with low supplies of oseltamivir may suffer higher death rates if swine flu becomes more lethal outside of Mexico.
References
- Yeung, Y.-Y., Hong, S. and Corey, E.J. (2006) A short enantioselective pathway for the synthesis of the anti-influenza neuramidase inhibitor oseltamivir from 1,3-butadiene and acrylic acid. J. Am. Chem. Soc. 128, 6310–6311.
- Fukuta, Y., Mita, T., Fukuda, N., Kanai, M. and Shibasaki, M. (2006) De novo synthesis of Tamiflu via a catalytic asymmetric ring-opening of meso-aziridines with TMSN3 J. Am. Chem. Soc. 128, 6312–6313.
- Mita, T., Fukuda, N., Roca, F.X., Kanai, M. and Shibasaki, M. (2007) Second generation catalytic asymmetric synthesis of Tamiflu: allylic substitution route. Org. Lett. 9, 259–262.
- Satoh, N., Akiba, T., Yokoshima, S. and Fukuyama., T. (2007) A practical synthesis of (–)-oseltamivir Angew. Chem. Int. Ed. 46, 5734–5736.
- Trost, B.M. and Zhang, T. (2008) A concise synthesis of (–)-oseltamivir Angew. Chem. Int. Ed. 47, 1–4.
29 April 2009, Ethyl formate
Ethyl formate (CHEBI:52342), a low-molecular-weight volatile compound produced by many fruits and vegetables, has an important role as a flavour and aroma component. It has also been used as a fumigant possessing insecticidal and fungicidal properties [1]. The compound can potentially degrade to biogenic levels in the tissues of treated commodities in contrast to conventional chemicals which can persist as residues in food products. Ethyl formate was evaluated as a fumigant for stored grain as a potential alternative to the ozone-depleting fumigant methyl bromide (CHEBI:39275) and to phosphine (CHEBI:30278), which is under pressure owing to the development of strong resistance in stored grain insects [2]. Recently, astronomers have found ethyl formate in a giant dust cloud at the heart of the Milky Way [3]. While scouring their data, the team also found evidence for the lethal chemical propyl cyanide (CHEBI:51937) in the same cloud. The two molecules, whose sizes are comparable to the simplest amino acid, glycine (CHEBI:15428), are the largest yet discovered in deep space. Finding amino acids in interstellar space would raise the possibility of life emerging on other planets as they are one of the building blocks of life.References
- Simpson, T., Bikoba, V. and Mitcham, E.J. (2004) Effects of ethyl formate on fruit quality and target pest mortality for harvested strawberries. Postharvest Biology and Technology 34, 313–319.
- Haritos, V.S., Damcevski, K.A. and Dojchinov, G. (2006) Improved efficacy of ethyl formate against stored grain insects by combination with carbon dioxide in a 'dynamic' application. Pest Manag. Sci. 62, 325–333.
- Galaxy's centre tastes of raspberries and smells of rum, say astronomers. guardian.co.uk. Tuesday 21 April 2009
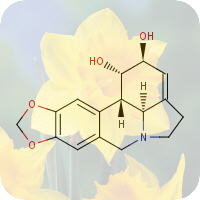
25 March 2009, lycorine
Daffodil is the common English name for Narcissus, the botanic name of a genus of mainly spring-flowering bulbs in the family Amaryllidaceae. Daffodil bulbs, often confused with onions, contain lycorine (CHEBI:6601), a highly poisonous crystalline alkaloid which might be lethal when ingested in certain quantities. Traditionally, lycorine has been obtained from plant extracts [1] but its total synthesis was recently accomplished using chiral ligand-controlled asymmetric cascade conjugate addition methodology [2]. Several studies have highlighted the capacity of lycorine to inhibit protein synthesis, most probably by acting at the level of termination [3]. Other published articles suggest a role as an inhibitor of ascorbic acid biosynthesis [4]. The most recent studies also indicate a possible role of lycorine as an anticancer drug bearing apoptosis-mediated antiprolifereative effects [5–7].References
- Heliang, F., Ning, W. and Xuanwang, H. (2003) Method for separating lycorine from plant extract. Patent CN1611504.
- Yamada, K.I., Yamashita, M., Sumiyoshi, T., Nishimura, K. and Tomioka, K. (2009) Total synthesis of (–)-lycorine and (–)-2-epi-lycorine by asymmetric conjugate addition cascade. Org. Lett. Publication Date (Web): February 25, 2009/DOI: 10.1021/ol9003564 .
- Vrijsen, R., Vanden Berghe, D.A., Vlietinck, A.J. and Boeyé, A.(1986) Lycorine: a eukaryotic termination inhibitor? J. Biol. Chem. 261, 505–507.
- Arrigoni, O., Arrigoni Liso, R. and Calabrese, G. (1975) Lycorine as an inhibitor of ascorbic acid biosynthesis. Nature 256, 513–514.
- Liu, J., Hu, W.-X., He, L.-F., Ye, M. and Li, Y. (2004) Effects of lycorine on HL-60 cells via arresting cell cycle and inducing apoptosis. FEBS Lett. 578, 245–250.
- Liu, X.-S., Jiang, J., Jiao, X.-Y., Wu, Y., Lin, J.-H. and Cai, Y.-M. (2008) Lycorine induces apoptosis and down-regulation of Mcl-1 in human leukemia cells. Cancer Lett. 274, 16–24 .
- Evidente, A., Kireev, A.S., Jenkins, A.R., Romero, A.E., Steelant, W.F., Van Slambrouck, S. and Kornienko, A. (2009) Biological evaluation of structurally diverse Amaryllidaceae alkaloids and their synthetic derivatives: discovery of novel leads for anticancer drug design. Planta Med. Publication Date (Web): February 23, 2009/DOI: 10.1055/s-0029-1185340.
25 February 2009, N'-bis(2,3-dihydroxybenzoyl)-N''-hexanoyltren
There is much interest in identifying compounds that transport anions, especially chloride (Cl–), across lipid membranes. Reports that catechol binds Cl– in organic solvents [1] have inspired Berezin and Davis [2] to use catechols for the synthesis of novel anion transporters. In a first step, two 2,3-dihydroxybenzoic acid residues were attached to a tris(2-aminoethyl)amine (tren) scaffold. In a second step, a hexanoyl group was linked to tren's third amino group to yield N,N'-bis(2,3-dihydroxybenzoyl)-N''-hexanoyltren (CHEBI:51950). The authors show that the selectivity of ionic permeability across the membrane in the presence of this bis-catechol compound follows a Hofmeister series [3] with the anion influx rate decreasing in the order ClO4– > I– > NO3– > Br– > Cl–. The authors also have observed a non-linear dependence of the rate constant for anion transport on the concentration of the transporter. The authors suggest that self-association of the bis-catechol transporter in the membrane increases the anion permeability across the bilayer without requiring complete dehydration of the transported anion. While bacterial siderophores and synthetic analogues use catecholates to transport iron(3+) ions across membranes, this is the first report of catechols facilitating transmembrane transport of anions.References
- Winstanley, K.J., Sayer, A.M. and Smith, D.K. (2006) Anion binding by catechols — an NMR, optical and electrochemical study. Org. Biomol. Chem. 4, 1760–1767.
- Berezin, S.K. and Davis, J.T. (2009) Catechols as membrane anion transporters. J. Am. Chem. Soc. 131, 2458–2459.
- Hofmeister, F. (1888) Zur Lehre von der Wirkung der Salze. Arch. Exp. Pathol. Pharmacol. 24, 247–260.
28 January 2009, Cucurbit[7]uril
In 1905, Behrend, Meyer and Rusche described the reaction between glycoluril and formaldehyde which yielded a cross-linked amorphous polymer. Treatment of this substance with hot concentrated sulfuric acid produced a crystalline precipitate [1]. Some 75 years after the Behrend et al. paper, the crystal structure of this compound (now known as cucurbit[6]uril or CB[6]) was solved by Freeman et al. The footnote to their paper [2] says: "The trivial name cucurbituril is proposed because of a general resemblance of 2 to a gourd or pumpkin (family Cucurbitaceae), and by devolution from the similarly named (and shaped) component of the early chemists' alembic."Now, the term cucurbituril refers to any macrocycle consisting of repeating cis-glycoluril residues linked by methylene groups, with the number of repeated units in the macrocycle denoted by the "n" in the cucurbit[n]uril (CB[n]) name. By virtue of their shape, cucurbiturils can form stable inclusion compounds and thus are now employed for molecular recognition, self-assembly, and nanotechnology [3]. In addition to that of CB[6], the structures of CB[5], CB[7], CB[8] and CB[10] have been determined. Unlike CB[6] or CB[8], which are sparingly soluble in water, CB[7] (CHEBI:51434) has a moderate solubility in water which prompted Jeon et al. to study its inclusion of drugs and explore its potential as a drug carrier [4]. CB[7] forms a stable 1:1 complex with the anticancer drug oxaliplatin in aqueous solution. The authors suggest that the encapsulation of the drug not only increases the stability of the drug, but also reduces the unwanted side effects of oxaliplatin.
References
- Behrend, R., Meyer, E. and Rusche, F. (1905) Ueber Condensationsproducte aus Glycoluril und Formaldehyd. Liebigs Ann. Chem. 339, 1–37.
- Freeman, W.A., Mock, W.L. and Shih, N.-Y. (1981) Cucurbituril. J. Am. Chem. Soc. 103, 7367–7368.
- Lagona, J., Muchophadyay, P., Chakrabarti, S. and Isaacs, L. (2005) The cucurbit[n]uril family. Angew. Chem. Int. Ed. 44, 4844–4870.
- Jeon, Y.J., Kim, S.-Y., Ko, Y.H., Sakamoto, S., Yamaguchi, K. and Kim, K. (2005) Novel molecular drug carrier: encapsulation of oxaliplatin in cucurbit[7]uril and its effects on stability and reactivity of the drug. Org. Biomol. Chem. 3, 2122–2125.
2008
17 December 2008, Epicocconone
The ascomycete fungus Epicoccum nigrum is known to be a source of natural fluorescent compounds. One of them, epicocconone (CHEBI:51226) [1], also known as "Deep Purple" and "Lightning Fast", is a water-soluble, low molecular mass molecule based on a polyketide skeleton. It has a weak green fluorescence in water (λem = 520 nm), which shifts to red (λem = 610 nm) with a concomitant increase in quantum yield in the presence of a protein. Its cell permeability, low cytotoxicity and large Stokes' shift make it a highly useful fluorochrome for live cell imaging [2]. Recently, a epicocconone-based assay that allows the rapid and accurate determination of protease enzyme kinetic parameters (KM, Vmax) as well as inhibition constants through the measurement of fluorescence anisotropy on the actual protease substrates [3] has been developed. This novel assay will be suitable for high-throughput applications in drug development and biotechnology.
References
- Bell, P.J.L. and Karuso, P. (2003) Epicocconone, a novel fluorescent compound from the fungus Epicoccum nigrum. J. Am. Chem. Soc. 125, 9304–93045.
- Choi, H.-Y., Veal, D.A. and Karuso, P. (2005) Epicocconone, a new cell-permeable long Stokes' shift fluorescent stain for live cell imaging and multiplexing. J. Fluoresc. 16, 475–482.
- Cleemann, F. and Karuso, P. (2008) Fluorescence anisotropy assay for the traceless kinetic analysis of protein digestion. Anal. Chem. 80, 4170–4174.
26 November 2008, Irgarol 1051
Biologically active triazine compounds have been extensively used as herbicides for over five decades [1]. Their generally high toxicity and persistence have aroused growing environmental and public health concerns. In recent years, Irgarol 1051 (CHEBI:5962), a highly specific methylthioltriazine compound which inhibits photosynthesis, has been used in long-life antifouling coatings for marine applications to prevent the growth of algae [2]. The degradation and transformation of Irgarol-1051 in natural coastal seawater has been studied [3] and two major degradation processes have been identified: microbial metabolism and photolysis. Irgarol 1051 main degradation product, which is biologically less toxic, is known as M1 or GS265751 (CHEBI:51079). A number of other transformation products, such as M2 (CHEBI:51112) and M3 (CHEBI:51083), have been identified; however, only the descyclopropyl transformation product M1 is being consistently found in natural waters [4]. Recently, the occurrence and persistence of Irgarol 1051 and its main metabolite in the coastal waters of Southern England were researched. The results indicated organic rich sediments and paint residues as major sites of storage for Irgarol 1051, and that Irgarol 1051 may be classified as a persistent organic pollutant due to its long half-life [5].
References
- Lam, K.-H., Cai, Z., Wai, H.-Y., Tsang, V.W.-H., Lam, M.H.-W., Cheung, R.Y.-H., Yu, H. and Lam, P.-K. (2005) Identification of a new Irgarol-1051 related s-triazine species in coastal waters. Environmental Pollution 136, 221-230.
- Ciba® IRGAROL® 1051 factsheet.
- Lam, K.-H., Lei, N.-Y., Tsang, V.W.-H., Cai, Z., Leung, K.M.Y. and Lam, M.H.-W. A mechanistic study on the photodegradation of Irgarol-1051 in natural seawater. Mar. Pollut. Bull., in press.
- Lam, K.-H., Wai, H.-Y., Leung, K.M.Y., Tsang, V.W.-H., Tang, C.-F., Cheung, R.Y.-H. and Lam, M.H.-W. (2006) A study of the partitioning behavior of Irgarol-1051 and its transformation products. Chemosphere 64, 1177-1184.
- Zhou, J.L. (2008) Occurrence and persistence of antifouling biocide Irgarol 1051 and its main metabolite in the coastal waters of Southern England. Sci. Total. Environ. 406, 239-246.
29 October 2008, PP121
A pyrazolopyrimidine, known simply as PP121 (CHEBI:50915), has been found to block simultaneously two key enzymes involved in the growth of cancer cells. In a recent publication [1], Kevan Shokat, of the University of California at Berkeley, and collaborators report that PP121 is highly effective in inhibiting both oncogenic tyrosine kinases and phosphatidylinositol-3-OH kinases [PI(3)Ks], two protein families that are currently among the most intensely pursued cancer drug targets. The enzymes are involved in cell signalling pathways, alterations of which are involved in many types of cancer. This example of dual inhibition owes its effectiveness to the ability of the molecule to flex around the single bond connecting its pyrazolopyrimidine and pyrrolopyridine ring systems. Thus the molecule is able to change its shape slightly, enabling it to occupy a hydrophobic pocket in both enzyme classes. The researchers believe that the rational design approach employed by them demonstrates the possibility to identify potent, selective and drug-like molecules capable of targeting these two classes of oncogenic kinases, and in the long term to providing routes to more effective therapy for many cancers.
Reference
- Apsel, B., Blair, J.A., Gonzalez, B., Nazif, T.M., Feldman, M.E., Aizenstein, B., Hoffman, R., Williams, R.L., Shokat, K.M. and Knight, Z.A. (2008) Targeted polypharmacology: discovery of dual inhibitors of tyrosine and phosphoinositide kinases. Nature Chem. Biol. 4, 691–699.
24 September 2008, Dronedarone
Atrial fibrillation (AF) is the most common form of cardiac arrhythmia (abnormal heart rhythm). Risk increases with age, with 8% of people over 80 having AF. Historically, the most effective treatment for this disorder has been either oral or intravenous administration of the iodinated benzofuran derivative amiodarone (CHEBI:2663), a procedure which has been used safely in patients with even advanced heart failure. However the use of amiodarone has been limited by cumulative and often irreversible organ toxicity, especially in younger patients. In an effort to provide equivalent efficacy and safety with less toxicity, dronedarone (CHEBI:50659) [2], a pharmacologically related benzofuran derivative lacking the iodine moieties, has been developed [1] and subjected to clinical trials. The U.S. Food and Drug Administration (FDA) assigned priority review status to dronedarone in August 2008 following the exciting results of the ATHENA study [3], a large randomized trial which showed a significant decreased risk of cardiovascular hospitalizations or death from any cause in patients with AF treated with dronedarone. Moreover, data presented at the European Society of Cardiology Congress in September [4] suggests that dronedarone could also reduce the risk of stroke for certain elderly patients with AF.
References
- Laughlin, J.C. and Kowey, P.R. (2008). Dronedarone: A New Treatment for Atrial Fibrillation. J. Cardiovasc. Electrophysiol. Published Online: 13 Aug 2008.
- Press release Sanofi-Aventis. Paris, France 8 August 2008.
- Hohnloser, S.H., Connolly, S.J., Crijns, H.J., Page, R.L., Seiz, W. and Torp-Petersen, C. (2007) Rationale and design of ATHENA: A placebo-controlled, double-blind, parallel arm trial to assess the efficacy of dronedarone 400 mg bid for the prevention of cardiovascular hospitalization or death from any cause in patients with atrial fibrillation/atrial flutter. J. Cardiovasc. Electrophysiol. 19, 69-73.
- Connolly, S.J. (2008) ATHENA: The effect of dronedarone on cardiovascular outcomes and stroke in patients with atrial fibrillation. European Society of Cardiology Congress 2008; Munich, Germany. Clinical trials update 3.
3 September 2008, Putrebactin
Siderophores are natural ferric chelators synthesised and excreted by microorganisms. Under aerobic conditions, iron exists mostly in oxidation state III (ferric) and tends to form highly insoluble minerals. Therefore, many microorganisms depend on siderophores for iron uptake. Putrebactin (CHEBI:50432) is a macrocyclic siderophore produced by the marine bacterium Shewanella putrefaciens [1]. It is derived from N-hydroxy-N-succinylputrescine (HSP) and is structurally similar to desferrioxamine E. It was recently shown that desferrioxamine siderophore synthetase DesD catalyses ATP-dependent trimerisation–macrocyclisation of N-hydroxy-N-succinylcadaverine to desferrioxamine E [2]. A BLAST similarity search identified a gene called pubC within a conserved five-gene cluster in several Shewanella species that encodes a homologue of DesD [1]. It was suggested that pubABC encodes the enzymes required to assemble putrebactin from putrescine, with PubA catalysing the dioxygen and FADH2-dependent hydroxylation of putrescine to N-hydroxyputrescine, PubB catalysing its succinyl-CoA-dependent succinylation to HSP, and PubC catalysing ATP-dependent dimerisation of HSP to give pre-putrebactin and its subsequent macrocyclisation to putrebactin. The pubA, pubB and pubC genes are homologous to the alcA, alcB and alcC genes, respectively, which are involved in the biosynthesis of alcaligin, a dihydroxylated analogue of putrebactin isolated from Bordetella species [3].
References
- Kadi, N., Arbache, S., Song, L., Oves-Costales, D. and Challis, G.L. (2008) Identification of a gene cluster that directs putrebactin biosynthesis in Shewanella species: PubC catalyzes cyclodimerization of N-hydroxy-N-succinylputrescine. J. Am. Chem. Soc. 130, 10458–10459.
- Kadi, N., Oves-Costales, D., Barona-Gomez, F. and Challis, G.L. (2007) A new family of ATP-dependent oligomerization-macrocyclization biocatalysts. Nature Chem. Biol. 3, 652–656.
- Challis, G.L. (2005) A widely distributed bacterial pathway for siderophore biosynthesis independent of nonribosomal peptide synthetases. ChemBioChem 6, 601–611.
30 July 2008, 3,3'-Diindolylmethane
3,3'-Diindolylmethane (DIM;
References
- Le, H.T., Schaldach C.M., Firestone G.L. and Bjeldanes L.F. (2003) Plant-derived 3,3'-diindolylmethane is a strong androgen antagonist in human prostate cancer cells. J. Biol. Chem. 278, 21136–21145.
- Xue, L., Pestka, J.J., Li, M., Firestone, G.L. and Bjeldanes, L.F. (2008) 3,3'-Diindolylmethane stimulates murine immune function in vitro and in vivo. J. Nutr. Biochem. 10, 336–344.
- Stresser, D.M., Bailey, G.S., Williams, D.E. and Bjeldanes, L.F. (1995) The anticarcinogen 3,3'-diindolylmethane is an inhibitor of cytochrome P-450. J. Biochem. Toxicol. 10, 191–201.
- Sanderson, J.T., Slobbe, L., Lansbergen, G.W.A., Safe, S. and van den Berg, M. (2001) 2,3,7,8-Tetrachlorodibenzo-p-dioxin and diindolylmethanes differentially induce cytochrome P450 1A1, 1B1, and 19 in H295R human adrenocortical carcinoma cells. Toxicol. Sci. 61, 40–48.
25 June 2008, Motexafin gadolinium
Nuclear magnetic resonance imaging (MRI) is a non-invasive, non-radiative medical imaging method of growing clinical importance. Unfortunately, the difference in MRI signal between diseased and healthy tissues is often small. To enhance the visualization of the area of interest, paramagnetic contrast agents are often used, such as gadolinium compounds. "Texaphyrins" (for "Texas-size porphyrins") is the trivial name of a class of synthetic expanded porphyrins first prepared in 1988 by Jonathan Sessler and co-authors at the University of Texas at Austin [1]. The texaphyrins form stable coordination complexes with large metal cations such as lanthanoids. One such complex, the texaphyrin gadolinium(3+) complex known as motexafin gadolinium (CHEBI:50161; MGd), has been extensively studied as a potential MRI-detectable enhancer of X-ray cancer therapy. In addition, it has been found that in cancer cells MGd generates reactive oxygen species such as superoxide and induces apoptosis. In vitro studies show that MGd enhances tumour cell cytotoxicity with both ionizing radiation and a number of chemotherapeutic agents [2]. The California-based company Pharmacyclics, Inc. has been developing MGd (brand name Xcytrin®) for use in combination with radiation therapy for treatment of brain metastases from lung cancer. According to the company, "MGd has been evaluated in clinical trials involving over 1000 patients and has been found to be generally well-tolerated, without the usual side-effects seen with standard chemotherapy" [3]. Earlier this month, Pharmacyclics announced final data from a Phase 1/2 study showing a high complete response rate in patients with multiple recurrent non-Hodgkin's lymphoma who were treated with MGd in combination with an approved antibody-targeted radiation therapy.
References
- Sessler, J.L., Murai, T., Lynch, V. and Cyr, M. (1988) An "expanded porphyrin": the synthesis and structure of a new aromatic pentadentate ligand. J. Am. Chem. Soc. 110, 5586–5588.
- Magda, D. and Miller, R.A. (2006) Motexafin gadolinium: a novel redox active drug for cancer therapy. Semin. Cancer Biol. 16, 466–476.
- Motexafin Gadolinium: Cancer Treatment product candidate.
28 May 2008, (–)-α-Thujone
Originally formulated in Switzerland, absinthe became most popular in 19th century France. Many creative artists had their lives touched by absinthe, among them Toulouse-Lautrec, Oscar Wilde, Picasso and Vincent van Gogh. Absinthe was classically manufactured from dried wormwood (Artemisia absinthium), anise and fennel, which were steeped overnight in 85% (by volume) ethanol [1]. Popular lore has it that absinthe causes hallucinations, epileptic-like attacks, and bouts of madness for those who drink it [2]. Convulsions resembling epilepsy were observed in humans and induced in animals with toxic doses of absinthe. The essential oils were first implicated, then specifically wormwood, and finally one compound, the monoterpene (–)-α-thujone (CHEBI:9577). Despite (or maybe precisely because of) its popularity, absinthe was cast as a dangerous and addictive psychoactive drug. Between 1905 and 1913 Belgium, Switzerland, the United States, and Italy banned absinthe [1]. The bans were surprising, given that they were not supported by any scientific evidence that absinthe contained enough thujone or other compounds that could lead to "absinthism". An international research effort now offers firm scientific evidence that thujone is not the culprit behind those effects [3]. The authors conclude that the thujone concentration of preban (prior to 1915) absinthe was generally overestimated in the past. Analysis of postban (1915–1988) and modern commercial absinthes (2003–2006) shows that the encompassed thujone ranges of all absinthes are similar, disproving the supposition that a fundamental difference exists between preban and modern absinthes manufactured according to historical recipes. As the authors note, 'all things considered, nothing besides ethanol was found in the absinthes that was able to explain the syndrome "absinthism"'.
References
- Strang, J., Arnold, W.N. and Peters, T. (1999) Absinthe: what's your poison? BMJ 319, 1590-1592.
- Ritter, S. (2008) Absinthe myths finally laid to rest. C&EN 86, 42–43.
- Lachenmeier, D.W., Nathan-Maister, D., Breaux, T.A., Sohnius, E.M., Schoeberl, K. and Kuballa, T. (2008) Chemical composition of vintage preban absinthe with special reference to thujone, fenchone, pinocamphone, methanol, copper, and antimony concentrations. J. Agric. Food Chem. 56, 3073–3081.
30 April 2008, 2-deoxy-2-(18F)fluoro-α-D-glucose
Positron emission tomography (PET) is one of the most sensitive molecular imaging techniques today. PET imaging agents are labelled with radionuclides which decay by the emission of a positron (β+), the antimatter counterpart of an electron. After being emitted from the nucleus, the positron travels a short distance in the surrounding matter before it annihilates with an electron. The annihilation produces two γ-rays, which are emitted simultaneously in opposite directions and are then detected by an array of surrounding detectors. Most of the PET imaging probes applied in neurological research have been labelled with either 11C or 18F.
References
- Ametamey, S.M., Honer, M. and Schubiger, P.A. (2008) Molecular imaging with PET. Chem. Rev., in press.
- Kopka, K., Schober, O. and Wagner, S. (2008) 18F-labelled cardiac PET tracers: selected probes for the molecular imaging of transporters, receptors and proteases. Basic Res. Cardiol. 103, 131–143.
26 March 2008, (2-trans,6-trans)-farnesol
Nutria, or coypu (Myocastor coypus), are very large (5–9 kg) rat-like animals, originally introduced into North America, Europe, Asia and Africa from South America for their fur, but which are currently ravaging wetlands across Louisiana and the coast of the Gulf of Mexico. Because of their ability to reproduce at rapid speed (a female may have 3 litters per year of up to 13 young per litter), they are unwieldy to control if released into the wild. Now, Professor Athula Attygalle and his team at the Stevens Institute of Technology, New Jersey, in collaboration with groups at Cornell University and the University of Iowa, have identified several volatile compounds, including terpenoids, fatty alcohols, fatty acids and some of their esters, in the anal scent glands of the species. Using gas chromatographic retention times as well as electron-ionization (EI) and chemical-ionization (CI) mass spectrometry, they have identified the major terpenoid constituents as (2-trans,6-trans)-farnesol [CHEBI:16619; otherwise known as (E,E)-farnesol] and its esters. (2-trans,6-trans)-Farnesol is a sesquiterpene alcohol which is also present in many essential oils such as citronella, neroli, cyclamen, lemon grass, tuberose, rose, musk, balsam and tolu. It is also used in perfumery to emphasize the odours of sweet floral perfumes. By incorporating this compound and its esters into lures as bait, Professor Attygalle's discovery offers an environmentally friendly solution to the problem of removing nutria from sensitive coastal zones and marshlands without detrimentally affecting other species.
Reference
- Lee, H., Finckbeiner, S., Yu, J.S., Wiemer, D.F., Eisner, T. and Attygalle, A.B. (2007) Characterization of (E,E)-farnesol and its fatty acid esters from anal scent glands of nutria (Myocastor coypus) by gas chromatography–mass spectrometry and gas chromatography–infrared spectrometry. J. Chromatogr., A 1165, 136–143.
27 February 2008, Pseudocoenzyme B12
Sixty years ago, a crystalline compound, which provided a cure for a previously fatal disease pernicious anaemia, was isolated from liver. This compound has become known as vitamin B12 [1]. In the same year, Dorothy Crowfoot Hodgkin began her attempts to determine the structure of vitamin B12, and in 1964 was awarded the Nobel Prize in Chemistry "for her determinations by X-ray techniques of the structures of important biochemical substances", including penicillin and vitamin B12. Vitamin B12 is biosynthesised in certain bacteria, with animals obtaining their B12 from food or directly from resident B12-synthesising bacteria. All B12 cofactors are cobalt–corrinoid complexes. One of the cobalt axial ligands (designated α) is attached to the corrin nucleus via the nucleotide loop, while the other axial ligand (designated β) can vary. The coenzymatically active forms of the B12 vitamins possess an organic β-axial ligand, either methyl or 5'-deoxy-5'-adenosyl, attached via a physiologically rare carbon-to-cobalt bond. The "classic" B12 cofactors that contain 5,6-dimethylbenzimidazole (DMB) as α-axial ligands are called cobalamins. Pseudocoenzyme B12 (CHEBI:48572) is the most common example of a natural B12 cofactor with an α-axial ligand other than DMB – in this case, adenine [2]. The bacterium Salmonella enterica produces the corrin ring only under anaerobic conditions, but it can form B12 coenzymes aerobically by importing a corrinoid precursor, such as cobinamide (Cbi), and adding appropriate axial ligands. Wild-type S. enterica can use ethanolamine as an aerobic source of carbon and energy in the presence of vitamin B12. It also can synthesise vitamin B12 only when both Cbi and DMB are provided. Anderson et al. [3] have recently demonstrated that mutants of S. enterica isolated for their ability to degrade ethanolamine without added DMB can convert Cbi into pseudo-B12 cofactors. The mutations cause an increase in the level of free adenine and install adenine as cobalt α-ligand instead of DMB. Addition of DMB to these mutants stops the synthesis of pseudo-B12 cofactors and B12 cofactors are produced instead. Wild-type cells make pseudo-B12 cofactors during aerobic growth on propanediol and Cbi, and can use pseudo-B12 for all of their corrinoid-dependent enzymes.
References
- Rickes, E.L., Brink, N.G., Koniuszy, F.R., Wood, T.R. and Folkers, K. (1948) Crystalline vitamin B12. Science 107, 396–397.
- Taga, M.E. and Walker, G.C. (2008) Pseudo-B12 joins the cofactor family. J. Bacteriol. 190, 1157–1159.
- Anderson, P.J., Lango, J., Carkeet, C., Britten, A., Kräutler, B., Hammock, B.D. and Roth, J.R. (2008) One pathway can incorporate either adenine or dimethylbenzimidazole as an α-axial ligand of B12 cofactors in Salmonella enterica. J. Bacteriol. 190, 1160–1171.
30 January 2008, Triclocarban
The urea derivative triclocarban (TCC; CHEBI:48347) is an antibacterial and antifungal compound which for 45 years has been added in the USA and Europe to household and personal care products including bars of soap, body washes, cleansing lotions, wipes and detergents. An estimated 45,000 kg of the product are imported annually for the US market alone. The mechanism of its action involves inhibition of the enzyme enoyl-[acyl-carrier-protein] reductase (NADH) (EC 1.3.1.9), a key enzyme of the type II fatty acid synthase (FAS-II) system. Recently, a new UC Davis study, led by emeritus professor Bill Lasley, an expert on reproductive toxicology, has raised concerns by demonstrating that triclocarban can alter hormonal activity in rats and human cells in the laboratory. The study identified two key effects: in human cells triclocarban increased gene expression that is normally regulated by testosterone; and when male rats were fed triclocarban, testosterone-dependent organs such as the prostate gland grew abnormally large. Also, the mechanism by which triclocarban acts, i.e. by increasing hormone effects, is novel, as all previous studies of endocrine disruptors have found that these generally act by blocking or decreasing hormone effects. Lasley believes that the findings may lead eventually to an explanation for rises in some previously described reproductive problems that to date have been difficult to understand.
Reference
- Chen, J., Ahn, K.C., Gee, N.A., Mohamed, M.I., Duleba, A.J., Zhao, L., Gee, S.J., Hammock, B.D. and Lasley, B.L. (2007) Triclocarban enhances testosterone action: A new type of endocrine disruptor? Endocrinology 149, 1173–1179.
2007
19 December 2007, Salinosporamide A
Salinosporamide A (CHEBI:48045), a chlorinated natural product from the marine bacterium Salinispora tropica, is a 20S proteasome inhibitor currently in phase 1 human clinical trials for the treatment of the bone marrow cancer multiple myeloma as well as solid tumours. It is around 500 times more active than its dechloro analogue salinosporamide B. In a recent publication by researchers in La Jolla, California [1], it was reported that salL, an 849-base-pair gene in the sal biosynthetic cluster, is required for in vivo chlorination of salinosporamide A. Their work has determined that SalL protein catalyses the displacement of L-methionine from the primary metabolite S-adenosyl-L-methionine (SAM; December 2005 ChEBI Entity of the Month) to generate the intermediate 5'-chloro-5'-deoxyadenosine by a nucleophilic substitution reaction. The mechanism has similarities to a known fluorinase FlA from the soil bacterium Streptomyces cattleya [2], which proceeds via a 5'-fluoro-5'-deoxyadenosine intermediate, and is in contrast to the oxidative mechanism that predominates in biological chorinations. Through its use of such an orthogonal biological chlorination mechanism, it is likely that SalL will prove valuable for the biosynthesis of halogenated small molecules.
References
- Eustáquio, A.S., Pojer, F., Noel, J.P. and Moore, B.S. (2008) Discovery and characterization of a marine bacterial SAM-dependent chlorinase. Nature Chem. Biol. 4, 69–74.
- Dong, C., Huang, F., Deng, H., Schaffrath, C., Spencer, J.B., O'Hagan, D. and Naismith, J.H. (2004) Crystal structure and mechanism of a bacterial fluorinating enzyme. Nature 427, 561–565.
28 November 2007, A,D-di-p-benzi[28]hexaphyrin(1.1.1.1.1.1)
The German astronomer and mathematician August Ferdinand Möbius (1790–1868) is best known for his discovery (in 1858) of a non-orientable two-dimensional surface with only one side, which is now known as the Möbius band. It was independently discovered the same year by another German mathematician, Johann Benedict Listing. 100 years after its discovery, the Möbius topology caught the imagination of chemists [1]. In the 1930s, Erich Hückel predicted that annulenes which contain 4n+2 mobile electrons should be stable (aromatic), and that those with 4n electrons should be unstable (antiaromatic). However, the Hückel rule is reversed for annulenes with a 180° twist (Möbius annulenes) [2]: chemical Möbius bands with 4n electrons are aromatic and those with 4n+2 electrons are antiaromatic. Recently, Stępień et al. [3] have demonstrated that a molecule can be dynamically switched between Hückel and Möbius topologies by changing the polarity of the solvent. This effect was observed in an expanded porphyrin containing two p-phenylene groups. The compound, A,D-di-p-benzi[28]hexaphyrin(1.1.1.1.1.1) (CHEBI:47035) has 28 (i.e. 4n) mobile electrons and is antiaromatic in non-polar solvents such as hexane, as expected from the Hückel rule. However in polar solvents such as chloroform and N,N-dimethylformamide, the molecule changes conformation from “Hückel” (two-sided) to “Möbius” (one-sided) and antiaromaticity is lost. The difference in conformation is brought about by a 90° twist in one of the p-phenylene groups, which can be either parallel to the other p-phenylene group (Hückel topology) or perpendicular (Möbius topology). The two conformers possess vastly different optical signatures: the hexane solution is green and the chloroform solution is blue. Have a virtual tour over the chemical Möbius band!
References
- Rzepa, H.S. (2005) Möbius aromaticity and delocalization. Chem. Rev. 105, 3697–3715.
- Herges, R. (2007) Aromatics with a twist. Nature 450, 36–37.
- Stępień, M., Latos-Grażyński, S., Sprutta, N., Chwalisz, P. and Szterenberg, L. (2007) Expanded porphyrin with a split personality: a Hückel-Möbius aromaticity switch. Angew. Chem. Int. Ed. 46, 7869–7873.
31 October 2007, capsaicin
Capsaicin (CHEBI:3374) is the active component of chilli peppers, plants belonging to the genus Capsicum. First isolated in 1816 but not synthesised in the laboratory until 1930, it is one of a class of related substances known as capsaicinoids. It is an irritant for mammals and produces a sensation of burning in tissues into which it comes into contact. Capsaicin is believed to be synthesised in the interlocular septa of chilli peppers by addition of a branched-chain to vanillylamine. Biosynthesis depends on the gene Pun1 which encodes a putative acyltransferase [1]. In a recent paper [2], Clifford Woolf and his co-workers at Harvard Medical School and Massachusetts General Hospital report that capsaicin can be used in combination with QX-314, a derivative of the commonly used local anaesthetic lidocaine, to produce an anaesthetic which can completely block pain without causing numbness or paralysis. On its own, QX-314 is unable to pass through cell membranes to block their electrical activity: the role of capsaicin is to open large pores called TRPV1 ion channels, which allow the QX-314 to pass through and selectively block the cells' activity. The treatment is thought to have great potential in improving pain treatment during childbirth, dental procedures and surgery.
References
- Stewart, C., Jr., Kang, B.-C., Liu, K., Mazourek, M., Moore, S.L., Yoo, E.Y., Kim, B.-D, Paran, I. and Jahn, M.M. (2005) The Pun1 gene for pungency in pepper encodes a putative acyltransferase. Plant J. 42, 675–688.
- Binshtok, A.M., Bean, B.P. and Woolf, C.J. (2007) Inhibition of nociceptors by TRPV1-mediated entry of impermeant sodium channel blockers. Nature 449, 607–610.
26 September 2007, (–)-geosmin
The chemical origin of the characteristic odour of soil was first investigated in 1891 by the French chemist and politician Marcellin Berthelot, considered by many to be one of the greatest chemists of all time. However, it was not until 1965 that the responsible agent, (–)-geosmin (CHEBI:46702), was first isolated in pure form and the structure assigned. Later investigations concluded that (–)-geosmin is generated, via germacradienol (CHEBI:46734), from the universal acyclic sesquiterpene precursor farnesyl diphosphate (FPP, CHEBI:17407) by an enzyme (UniProtKB Q9X839, IntEnz EC 4.2.3.22) that is encoded by the SCO6073 gene in the soil organism Streptomyces coelicolor A3(2). A recent study by David Cane and his team at Brown University, Rhode Island, has now shown unambiguously that the SCO6073 germacradienol-geosmin synthase of S. coelicolor is a bifunctional enzyme, in which it is the N-terminal half of the protein which converts FPP principally to geramacradienol, while the C-terminal half (previously thought to be catalytically inactive) then rebinds the germacradienol and catalyses its proton-initiated cyclisation to give the (–)-geosmin.
Reference
- Jiang, J., He, X. and Cane, D.E. (2007) Biosynthesis of the earthy odorant geosmin by a bifunctional Streptomyces coelicolor enzyme. Nature Chem. Biol. 3, 711–715.
29 August 2007, imatinib
Protein-tyrosine kinases represent an important class of drug targets, particularly in oncology and inflammation. In a recent study [1], a "chemical proteomics" methodology that enables the capturing of a defined sub-proteome (in this case, protein kinases) on a mixed kinase inhibitor matrix ("kinobeads"), and subsequent analysis by quantitative protein mass spectrometry, has been described. This methodology was applied to three drugs targeting the oncogenic BCR-ABL kinase, which induces chronic myelogenous leukaemia (CML). Quantitative profiling of the drug candidate bosutinib (SKI-606), which is currently undergoing clinical trials, and the marketed drugs imatinib (CHEBI:38918) and dasatinib in K562 cells confirms known targets including ABL and SRC family kinases. Unexpectedly, the receptor tyrosine kinase DDR1 and the metalloflavoprotein oxidoreductase NQO2 (EC 1.10.99.2) were also identified as potent novel targets of imatinib. The function of the "forgotten" enzyme NQO2 (discovered in 1961; re-discovered 36 years later!) is not known but it has been implicated in the detoxification of xenobiotics. It also has been shown to be inhibited by the anti-malarial drugs chloroquine and quinacrine as well as by the red wine component resveratrol [2]. Interestingly, a high expression of NQO2 is found in myeloid cells, which are also the target of imatinib in CML.
References
- Bantscheff, M. et al. (2007) A quantitative chemical proteomics approach reveals novel modes of action of clinical ABL kinase inhibitors. Nature Biotechnol. 25, 1035–1044.
- Vella, F., Ferry, G., Delagrange, P. and Boutin, J.A. (2005) NRH:quinone reductase 2: an enzyme of surprises and mysteries. Biochem. Pharmacol. 71, 1–12.
25 July 2007, water
It is a wet summer this year in Britain and so it may be an appropriate time to reflect on the unique chemical compound known as water (CHEBI:15377). Water was one of the classic "four elements" of antiquity. It was Antoine Lavoisier who showed in the late 18th century that water is composed of two elements, hydrogen and oxygen. Water covers >70% of the Earth's surface and is the only single chemical substance to give its name to a scientific discipline, that of hydrology. Water is the single most important molecule for life on Earth. Most biochemical reactions take place in aqueous solution; water also is a substrate of many of them (hydrolases, hydratases, photosystem II). Crystalline proteins contain significant amounts of water. Some proteins contain single-file water chains that are suggested to be able to conduct protons along their length ("proton wires"). Such proton wires were observed in X-ray structures of the photosynthetic reaction centre from Rhodobacter sphaeroides, bacteriorhodopsin, cytochrome f, cytochrome c oxidase and phosphatidylglycerophosphatase from Listeria monocytogenes [1 and references therein]. Water can play a structural function in proteins, as is shown by the recent X-ray structural determination of a cysteinless plant Kunitz-type protease inhibitor BbCI where a water-mediated hydrogen bond replaces the highly conserved disulfide bridge thought to be crucial for protein structure stabilisation [2].
References
- Kumaran, D., Bonanno, J.B., Burley, S.K. and Swaminathan, S. (2006) Crystal structure of phosphatidylglycerophosphatase (PGPase), a putative membrane-bound lipid phosphatase, reveals a novel binuclear metal binding site and two "proton wires". Proteins 64, 851–862.
- Hansen, D., Macedo-Ribeiro, S., Verissimo, P., Yoo Im, S., Sampaio, M.U. and Oliva, M.L.V. (2007) Crystal structure of a novel cysteinless plant Kunitz-type protease inhibitor. Biochem. Biophys. Res. Commun. 46, 735–740.
27 June 2007, lapatinib
Lapatinib (CHEBI:38636) is a quinazolinamine-based small molecule that potently inhibits the intracellular tyrosine kinase domains of the epidermal growth factor receptor (EGFR) and the human epidermal growth factor receptor 2 (HER2). In March 2007, lapatinib, in the form of its ditosylate monohydrate [Tykerb; GlaxoSmithKline (GSK)], was approved by the US Food and Drug Administration (FDA) for the treatment of patients with advanced or metastatic breast cancer whose tumours overexpress HER2 and who have received prior therapy including an anthracycline, a taxane and trastuzumab (Herceptin), the combination of trastuzumab with taxane-based chemotherapy being currently the standard first-line treatment for HER2-positive metastatic breast cancer. GSK also last year filed an application for the drug to the European Medicines Agency and expect approval for its use within Europe later this year. Financial analysts are predicting peak sales for lapatinib of $1.5 billion [Moy, B., Kirkpatrick, P., Kar, S. and Goss, P. (2007) Lapatinib. Nature Reviews Drug Discovery 6, 431–432].
30 May 2007, chlorophyll a
Photosystem I (PSI), a protein supercomplex that contains a reaction centre and light-harvesting complexes, is involved in oxygenic photosynthesis where it is a star performer in energy-gathering terms. It is the most efficient photochemical machine in nature, with almost every photon absorbed by the PSI complex being used to drive electron transport. In a recent issue of Nature, Nathan Nelson and his team at Tel Aviv University report the X-ray crystal structure of plant PSI from pea (Pisum sativum) and reveal that, along with a small number of phylloquinones, Fe4S4 clusters and carotenoids, this huge complex binds 168 chlorophyll molecules, with most of these being our Entity of the Month chlorophyll a (CHEBI:18230). Thus, even though chlorophyll is usually thought of as a "cofactor", the sheer bulk of chlorophylls in PSI makes us (ChEBIsts) look at it differently. One can say that PSI is mostly chlorophyll with some proteins wrapped around! [Amunts, A., Drory, O., Nelson, N. (2007) The structure of a plant photosystem I supercomplex at 3.4 Å resolution. Nature 447, 58–63].
25 April 2007, brussalexin A
Phytoalexins are antimicrobial compounds synthesised by plants when stressed which help the weakened plant to defend itself against infection. Soledade Pedras and her research team at the University of Saskatchewan have been using UV irradiation to stress Brussels sprouts (Brassica oleracea var. gemmifera) in order to discover new molecules and have now isolated from their experiments a remarkable new phytoalexin which they name brussalexin A (CHEBI:38130). The compound contains an allyl thiocarbamate group, a structure not previously seen in phytoalexins and whose formation cannot be explained using currently known pathways. Brussalexin A proves to be toxic towards several species of fungus, in particular Sclerotina sclerotiorum, which causes stem rot in many plant families. Pedras' group will now investigate how fungal pathogens break down brussalexin A, with their principal aim being to treat crops with compounds that inhibit these detoxification routes, thus allowing the plants' natural defence mechanisms to work for longer [Pedras, M.S.C., Zheng, Q.-A. and Sarwar, M.G. (2007) Efficient synthesis of brussalexin A, a remarkable phytoalexin from Brussels sprouts. Org. Biomol. Chem. 5, 1167–1169; see also the article "Stressed sprouts hit back" in Chemical Biology 2007, vol. 5.].
28 March 2007, cimicifoetiside A
Cimicifuga foetida is a herbaceous perennial plant native to China where it is used as an antipyretic and analgesic agent in traditional Chinese medicines. Alcoholic extracts from its rhizomes are now being imported into Western markets in order to meet an ever-increasing demand for its North American relative C. racemosa (known commonly as black cohosh, black bugbane or black snakeroot) which is widely used in the United States and Europe as a herbal dietary supplement for the relief of symptoms related to menopause. A collaboration between workers in Kunming, China, and Washington University in the US investigating the constituents of C. foetida has recently resulted in the isolation of a novel cycloartane-type triterpene glycoside which they have named cimicifoetiside A (CHEBI:37779), together with its 25-O-acetyl derivative, named cimicifoetiside B (CHEBI:37780). The compounds are interesting both structurally, in that they possess a relatively uncommon 2-O-acetyl α-L-arabinosyl unit, and physiologically, as they exhibit potent cytotoxicity against human breast cancer cells, suggesting the potential for further examination of these and other cycloartane triterpene glycosides from Cimicifuga spp. for the prevention or treatment of human breast cancers [Sun et al. (2007) Beilstein Journal of Organic Chemistry 3,3].
28 February 2007, beauvericin
Beauvericin (CHEBI:3000) is a cyclodepsipeptide of alternating L-N-methylphenylalanyl and D-α-hydroxyvaleryl residues. Well known as a mycotoxin affecting wheat and other cereal crops, it is produced by several Fusarium  species and has also been isolated from the entomopathogenic fungus Beauvaria bassiana . Beauvericin is capable of complexing both alkaline-earth metals and alkali-metal ions and transporting them through membranes in biological systems. New reports of its properties and usage constantly emerge, as exemplified by a very recent publication from two groups in Arizona in which inhibition by beauvericin of metastatic prostate cancer and breast cancer cells, as well as antiangiogenic activity, is demonstrated [Zhan et al. (2007) J. Nat. Prod. 70,227–232 ].
species and has also been isolated from the entomopathogenic fungus Beauvaria bassiana . Beauvericin is capable of complexing both alkaline-earth metals and alkali-metal ions and transporting them through membranes in biological systems. New reports of its properties and usage constantly emerge, as exemplified by a very recent publication from two groups in Arizona in which inhibition by beauvericin of metastatic prostate cancer and breast cancer cells, as well as antiangiogenic activity, is demonstrated [Zhan et al. (2007) J. Nat. Prod. 70,227–232 ].
31 January 2007, (–)-epigallocatechin 3-gallate
Catechins (CHEBI:23053) are a group of polyphenolic plant metabolites found extensively in extracts of green, unfermented, tea (Camellia sinensis ). The various health benefits attributed to green tea have until recently been thought to be linked to the antioxidant properties of its constituent catechins. However the use of green tea extract in oral hygiene for several centuries suggests that green tea may also have antibacterial activity. Roman Jerala and his colleagues at the National Institute of Chemistry in Ljubljana, Slovenia, have now, using heteronuclear 2D NMR and fluorescence spectroscopy, studied the properties of (–)-epigallocatechin 3-gallate (EGCG) (CHEBI:4806), the major catechin found in green tea. Their results show that ECGC inhibits the bacterial enzyme DNA gyrase by binding to the ATP binding site of one of its two subunits, known as gyrase B. This subunit catalyses ATP hydrolysis, providing the driving force for supercoiling of DNA, and has been previously investigated as a target for antibacterial drugs. Jerala believes that his findings with EGCG should allow pharmaceutical companies to design novel analogues which improve on EGCG's antibacterial activity without prohibitive side effects emerging [Gradišar, H., Pristovšek, P., Plaper, A. and Jerala, R. (2007) Green tea catechins inhibit bacterial DNA gyrase by interaction with its ATP binding site. J. Med. Chem. 50, 264–271 ; see also: Bradley, D. Tea for who? SpectroscopyNOW.com, January 15, 2007 ].
2006
20 December 2006, tangeretin
The tradition of putting a tangerine (Citrus nobilis var. Tangeriana) into the toe of a Christmas stocking is thought by some to represent the landing within stockings hung up to dry of dowry money which St Nicholas ('Santa Claus') tossed down a chimney in order to rescue several poor maidens from being sold into slavery. In more recent years, however, a more scientific importance of tangerines has emerged, namely that a valuable constituent of their peel (and that of some other citrus fruits) is tangeretin (CHEBI:9400), a polymethoxylated flavone which exhibits a wide range of pharmacological activity. In vitro studies have demonstrated antimutagenic, antiinvasive and antiproliferative effects, while animal research has suggested its potential as both a cholesterol-lowering agent and a protector against Parkinson's disease. More significantly perhaps is the potential of tangeretin as an anti-cancer agent, demonstrated by its inhibition of leukaemic HL-60 cell growth through induction of apoptosis [Hirano et al. (1995) Br. J. Cancer 72, 1380-1388 ], although it must be noted that in the presence of dietary tangeretin a neutralization of the tumour-inhibitory effect of the anti-cancer drug tamoxifen (CHEBI:9396) is observed [Bracke et al. (1999) J. Natl. Cancer Inst. 91, 354-359 ]. A very recent report [Takano et al. (2007) Am. J. Physiol. Cell Physiol. 292, C353–C361 ] highlights the ability of tangeretin and similar polymethoxyflavones to protect cells against endoplasmic reticulum (ER) stress.
1 November 2006, (R)-amygdalin
(R)-Amygdalin (CHEBI:17019; commonly known merely as 'amygdalin' or as 'Vitamin B17') is a by-product of the fruit industry. A naturally occurring cyanogenic glycoside, it is found in many food plants such as the kernels of apricots, almonds and apples and is used as a main constituent in commercial preparations of laetrile, a purported therapeutic agent. George John and Praveen Kumar Vemula from the City College of the City University of New York have now used an enzyme catalysis route to make amygdalin-based amphiphiles (molecules with parts which engage separately in hydrophilic and hydrophobic interactions) which showed unprecedented gelation properties in a wide range of solvents. Using further enzyme catalysis, the supramolecularly organised hydrogels thus formed were then employed as a successful drug-delivery vehicle for curcumin, a well known drug with anti-inflammatory and anti-cancer properties. The project is part of John's ongoing work on the design and development of new soft nanomaterials derived from plant/crop-based renewable feedstocks [John, G. and Vemula, P.K. (2006) Design and development of soft nanomaterials from biobased amphiphiles. Soft Matter 2, 909-914].
27 September 2006, ciguatoxin
The huge ladder-like polycyclic ether ciguatoxin (CHEBI:36467) is a marine toxin found in tropical fish. It is produced by an epiphytic dinoflagellate, Gambierdiscus toxicus, and transferred to herbiverous and carnivorous fish through the aquatic food chain. Ciguatoxin and its congeners are the principal causative agents of ciguatera poisoning, the symptoms of which include gastrointestinal, cardiovascular and neurological disorders which can last for many years. Ciguatoxin is one of the most lethal natural products known with a toxicity almost 300 times that of tetrodotoxin, the infamous compound found in pufferfish. Until now, detailed biological studies, as well as the development of therapeutic methods for ciguatera, have been hampered by the very limited supply of the natural product (the yield from 4000 kg of moray eels is only 0.35 mg). Professor Masayuki Inoue and his colleagues in the Hirama Resarch Group at Tohoku University, Sendai, have now published the first total synthesis of ciguatoxin and one of its hydroxylated derivatives, an achievement that will undoubtedly lead to the development of strategies to contol ciguatera food poisoning [M. Inoue et al. (2006) Total Synthesis of Ciguatoxin and 51-HydroxyCTX3C. J. Am. Chem. Soc. 1280, 9352-9354].
30 August 2006, 19-norandrosterone
19-Norandrosterone (CHEBI:36412) is a naturally produced steroid and a metabolite of the anabolic steroid nandrolone, which is still used illegally by some athletes to boost muscle mass. Evidence for nandrolone abuse is normally provided from analysis of urine samples for 19-norandrosterone, although current tests are unable to distinguish whether the norandrosterone is of synthetic or physiological origin (it is produced naturally in the adrenal glands, gonads and placenta). Researchers at the German Sport University Cologne and the Montreal Anti-doping Laboratory at the INRS-Institut Armand-Frappier have recently developed a technique called gas chromatography combustion isotope ratio mass spectrometry, which can distinguish between the two by analysing the ratio of 13C to 12C in the urine samples. The method can detect concentrations of 19-norandrosterone as low as 2 ng per ml of urine (the level currently allowed by the World Anti-Doping Agency) and may help to resolve cases where athletes have elevated levels of 19-norandrosterone in their urine but insist that they have never taken steroids [Hebestreit et al. (2006) Analyst 131, 1021-1026].
24 July 2006, ozone
Ozone (O3; CHEBI:25812), one of the most toxic inorganic compounds known, is present in low concentrations throughout the Earth's atmosphere. Ground-level ozone, formed by reaction of hydrocarbons and nitrogen oxides with sunlight, is an air pollutant with harmful effects on lung function. A current prolonged period of hot sunny weather across the UK, with daytime temperatures over 30 degrees Celsius, combined with a flow of air from continental Europe containing ozone precursor particulates, led during July of this year to the UK government department Defra issuing a Smog Warning. Levels of ozone across the UK are monitored by the Air Quality Archive, which also offers health advice to those who may be particularly sensitive to air pollution.
27 June 2006, C60 fullerene
C60 fullerene (CHEBI:33128; known also as buckminsterfullerene, footballene and soccerballene) was discovered in 1985 by Sir Harold Kroto in the UK and Richard E. Smalley and Robert F. Curl, Jr. in the USA. These three researchers shared The 1996 Nobel Prize in Chemistry for their discovery. C60 fullerene's atoms are bonded together into a highly symmetrical, hollow polygon structure with the same geometry as that of a football (soccerball). Named after the architect Richard Buckminster "Bucky" Fuller whose geodesic dome design is similar to the molecular structure of C60, these unique structures, also known as buckyballs, have led to an entirely new branch of chemistry. A recent review lists several interesting biological and pharmacological properties of C60 fullerene and derivatives, such as nitric oxide synthase inhibition, nitric oxide-scavenging activity, reactive oxygen species-generating activity, superoxide dismutase mimic properties, and HIV reverse transcriptase and RNA polymerase inhibition [Satoh, M. and Takayanagi, I. (2006) Pharmacological studies on fullerene (C60), a novel carbon allotrope, and its derivatives. J. Pharmacol. Sci. 100, 513–518]. The ChEBI team find it fitting that this 10th anniversary of the Fullerene Nobel Prize should coincide with the staging of the 2006 Football World Cup!
31 May 2006, porphyra-334
The mycosporine-like amino acid porphyra-334 (CHEBI:35671) was first isolated in 1979 from the red alga Porphyra tenera Kjelman. Morris Srebnik and his team at The Hebrew University of Jerusalem have now also isolated porphyra-334 from the aquatic bacterium Aphanizimenon flos-aquae and compared its UVA absorption with that of some commercial sun-care products. Their results show that porphyra-334 provides an equivalent of sun protection factor (SPF) 4 against UVA rays. Furthermore, it dissipates UV energy without creating any reactive species which might cause a phototoxic effect on living organisms, suggesting its possible further development as a commercial sunscreen [Torres, A., Enk, C.D., Hochberg, M. and Srebnik, M. (2006) Porphyra-334, a potential natural source for UVA protective sunscreens. Photochem. Photobiol. Sci. 5, 432–435].
26 April 2006, adenosine 5'-monophosphate 1-oxide
Royal jelly is a sticky, creamy white secretion produced by honey bees to feed the larvae of the colony until they develop to the desired rank. The larvae to develop into the queen will receive only royal jelly as their food source. Royal jelly has been claimed to have various beneficial effects on human health. It is a mixture of vitamins, amino acids, proteins, sugars and lipids, as well as the hero of this month, adenosine 5'-monophosphate 1-oxide (CHEBI:35483), or AMP 1-oxide. AMP 1-oxide was previously shown to inhibit the growth of tumor cells in culture as well as vaccinia virus replication by the blockade of translation of virus mRNAs. However, there have been no reports showing the distribution of adenosine 1-oxide derivatives in natural products, or the activity on the nervous system. Recently, Hattori and co-authors have shown that AMP 1-oxide is an active component of royal jelly that induces neuronal differentiation of pheochromocytoma PC12 cells. AMP 1-oxide induced neurite (process)-formation, inhibited cell growth, and facilitated neurofilament M expression, which suggests that AMP 1-oxide stimulates neuronal differentiation of PC12 cells similarly to nerve growth factor (NGF). [Hattori, N., Nomoto, H., Mishima, S., Inagaki, S., Goto, M., Sako, M. and Furukawa, S. (2006) Identification of AMP
22 March 2006, (25R)-5β-spirostan-1β,3α-diol
(25R)-5β-spirostan-1β,3α-diol (CHEBI:35370) is a previously unknown naturally occurring steroidal sapogenin which has been recently isolated from the bulbs of a southern African grassland lily, Ornithogalum tenuifolium. The steroid monomers stack to form one-dimensional chains that interlock with neighbouring polymers, with the overall structure of the natural product almost perfectly mimicking a man-made zip-fastener. It is the first-known zip-like structure to be composed of individual steroid molecules rather than the more usual coupled polymer chains such as those found in DNA [Munro et al. (2006) New J. Chem. 30, 197–207].
22 February 2006, apratoxin A
Apratoxins are marine cyanobacterial cyclodepsipeptides containing discrete polypeptide and polyketide domains. Apratoxin A (CHEBI:35212) demonstrates potent cytotoxicity against tumor cell lines through an unknown mechanism. In a recent communication, Luesch and co-authors used a functional genomics approach, including mRNA expression analysis and genome-wide arrayed cDNA overexpression, to elucidate the molecular basis for this activity. The authors conclude that apratoxin A mediates tumor cytotoxicity through the induction of cell cycle arrest and of apoptosis, which is at least partially initiated through antagonism of fibroblast growth factor (FGF) signalling [Luesch, H., Chanda, S.K., Raya, R.M., Dejesus, P.D., Orth, A.P., Walker, J.R., Izpisúa Belmonte, J.C. and Schultz, P.G. (2006) A functional genomics approach to the mode of action of apratoxin A. Nature Chemical Biology 2, 158–167].
25 January 2006, cis-bis(μ-acetato)[tetrakis(acetonitrile)]diaquadirhodium(Rh—Rh)(2+)
The dinuclear rhodium complex cis-bis(μ-acetato)[hexakis(acetonitrile)]dirhodium(Rh—Rh)(2+) (CHEBI:33863) exchanges its two axial acetonitrile ligands for solvent molecules in water to yield a diaqua complex, cis-bis(μ-acetato)[tetrakis(acetonitrile)]diaquadirhodium(Rh—Rh)(2+) (CHEBI:33894). It has recently been shown that upon irradiation with visible light, the diaqua complex exchanges two further acetonitrile ligands to yield a tetraaqua complex cis-bis(μ-acetato)[bis(acetonitrile)]tetraaquadirhodium(Rh—Rh)(2+) (CHEBI:33895), which is able to bind covalently to double-stranded DNA. The low cytotoxicity in the dark makes the diaqua complex a promising potential agent for photodynamic therapy [Lutterman, D.A., Fu, P.K.-L. and Turro, C. (2006) J. Am. Chem. Soc. 128, 738–739].
2005
7 December 2005, S-adenosyl-L-methionine
S-adenosyl-L-methionine (AdoMet; SAM; CHEBI:15414) is an important sulfonium intermediate in one-carbon metabolism, the 'active methyl' of the methionine being donated to an acceptor molecule by transmethylation with production of S-adenosyl-L-homocysteine. A recent communication reports the synthesis of analogues withextended allylic and propargylic chains replacing the methyl group and shows their suitability as cofactors for DNA methyltransferases, providing a new method for sequence-specific covalent derivatization of DNA.
26 October 2005, quinacrine
The human prion protein in its normal form, PrPc, plays an important role in maintaining several cell functions. The pathogenic form, PrPSc, has an essentially identical primary structure but is folded differently and displays different physiological interactions giving rise to prion diseases such as variant CJD. Quinacrine (CHEBI:8711), an acridine derivative formerly widely used as an antimalarial, is known to bind to PrPc prions and is currently undergoing human clinical trials through the British Medical Research Council to investigate its efficacy in combating such prion diseases.
22 September 2005, tomatine
The glycoalkaloid tomatine (CHEBI:9630) is a steroidalsaponin that occurs in the leaves of wild tomato plants where it inhibits the growth of various fungi and bacteria. Researchers at the University of East Anglia in the UK have recently published a one-pot synthetic route to the tetrasaccharide part of tomatine which should facilitate studies into understanding the ecological relationships between plants and fungi [Jones, N.A., Nepogodiev, S.A. and Field, R.A. (2005) Organic & Biomolecular Chemistry 3, 3201–3206).
31 August 2005, rofecoxib
Rofecoxib (CHEBI:8887), a cyclooxygenase (COX) inhibitor, was formerly marketed by Merck and Co. under the trade name VIOXX. Its specificity for only one form of the enzyme, COX-2, allowed it to reduce inflammation and pain while minimizing undesired gastrointestinal adverse effects, common with other nonsteroidal anti-inflammatory drugs. On August 19 2005 a US jury found Merck negligent in the death of a man who used it and awarded his widow $253.4m. Merck is to appeal against the decision. A view of the moleculeand some further reading are available here.
27 July 2005, ATTA-Eu3+
A dioxygen molecule in an excited singlet state, known as singlet molecular oxygen (1O2), reacts withmany kinds of biomolecules, such as DNA, proteins and lipids. Researchers at the Chinese Academy of Sciences have recently reported the synthesis and characterization of ATTA-Eu3+ (CHEBI:33025), the first europium(3+) chelate-based phosphorescence probe specific for time-resolved luminescence detection of 1O2. The structure of ATTA-Eu3+ is shown in the abstract of this report [Song, B., Wang, G. and Yuan, J. Chem. Commun., 2005, 3553–3555].
29 June 2005, pelargonidin 3-glucoside
The predominant pigment in strawberries (Fragaria sp.) is pelargonidin 3-glucoside (CHEBI:31967). One of three anthocyanins which contribute to the red colour inripe fruit, it is present at a concentration of around 900 μmol/kg and metabolised in humans principally viaformation of various pelargonidin monoglucuronic acid conjugates. A view of the molecule is shownhere.
25 May 2005, ellagic acid
Ellagic acid [CHEBI:4775] is a fused four-ring polyphenol found abundantly in various fruits, nuts and vegetables. It is active in antimutagenesis assays, and has been shown to inhibit chemically induced cancer in the lung, liver, skin and oesophagus of rodents, and TPA-induced tumour promotion in mouse. Studies suggest that the mechanisms of mutagenesis and carcinogenesis involve adduct formation with DNA, thus masking binding sites to be occupied by the mutagen or carcinogen. A representation of the molecule is available here.
27 April 2005, vancomycin
Vancomycin [CHEBI:28001], a glycopeptide isolated from Streptomyces orientalis, inhibits a specific step in the synthesis of the peptidoglycan layer in Staphylococcus aureusand Clostridium difficile. For over 40 years it has been used to kill bacteria when no other drug works and has become known as the "antibiotic of last resort". However, in recent years vancomycin-resistant enterococci (VRE) have emerged,prompting restrictions in the drug's use. A 3-D representation of the molecule is available here.
16 March 2005, cisplatin
Cisplatin [CHEBI:27899], the structure of which is incorporated into the ChEBI logo, is a square planar platinum complex widely used for the treatment of a variety of tumours. Its mode of action involves loss of its Cl− ligands and binding mainly to the N-7 atoms of a pair of guanine bases on either the same or adjacent strands of DNA, thus causing distortion of the DNA structure and inhibition of cell repair. A diagram showing such linking occurring between adjacent DNA strands is available here.
